Bulk RNA differential expression
Overview
Teaching: 60 min
Exercises: 120 minQuestions
How do I normalize a bulk RNA dataset?
How can I summarize the relationship between samples?
How can I compare expression based on biological or technical variables?
How can I visualize the results of this comparison?
How can I prepare differential expression results for input into downstream tools for biological insight?
How can I interpret the results of functional enrichment tools?
Objectives
Apply standard bulk workflow pre-processing methods such as DESeq2 normalization given a gene-sample count matrix.
Execute dimensional reduction (PCA) and corresponding visualization.
Understand design formulas and how to apply them to run differential expression.
Summarize differential expression (DE) results using a heatmap visualization as well as MA and volcano plots.
Interpret data visualizations from the above steps.
Use base R commands to output text files from DE results for input into functional enrichment (ORA/GSEA).
Run ORA/GSEA using a web-based tool, and describe the meaning of the resulting output.
Lecture portion
The workshop will include a lecture component of around one hour before the practical session later on.
Slides for this available here.
About this tutorial
This workflow is based on material from the rnaseqGene page on Bioconductor (link).
This data is from a published dataset of an RNA-Seq experiment on airway smooth muscle (ASM) cell lines. From the abstract:
“Using RNA-Seq, a high-throughput sequencing method, we characterized transcriptomic changes in four primary human ASM cell lines that were treated with dexamethasone - a potent synthetic glucocorticoid (1 micromolar for 18 hours).”
Citation:
Himes BE, Jiang X, Wagner P, Hu R, Wang Q, Klanderman B, Whitaker RM, Duan Q, Lasky-Su J, Nikolos C, Jester W, Johnson M, Panettieri R Jr, Tantisira KG, Weiss ST, Lu Q. “RNA-Seq Transcriptome Profiling Identifies CRISPLD2 as a Glucocorticoid Responsive Gene that Modulates Cytokine Function in Airway Smooth Muscle Cells.” PLoS One. 2014 Jun 13;9(6):e99625. PMID: 24926665. GEO: GSE52778
DOI link here.
Setting up to run this tutorial
Open Launcher.

Go to “R” under “Notebook”.

Press the “+” at the top to open another tab.
This time, go to “Other”, then “Terminal”.


Locating the data on the file system
This section will be run in the “Terminal” tab.
The data has already been downloaded, and is in CSV format for easy reading into R.
The main files we will be working with today are a gene x sample count matrix, and a metadata file with design information for the samples.
Data is available in this directory:
/data/RNA/bulk
If we list that path like so:
ls /data/RNA/bulk
We find the following files:
airway_raw_counts.csv.gz
airway_sample_metadata.csv
To list the files with their full paths, we can use *.
ls /data/RNA/bulk/*
/data/RNA/bulk/airway_raw_counts.csv.gz /data/RNA/bulk/airway_sample_metadata.csv
Bulk RNA differential expression workflow in R with DESeq2
The following will all be run in the R notebook tab.
However, let’s also leave the Terminal tab open for later.
Load libraries.
Load all the libraries you will need for this tutorial using the library command. Today we will load DESeq2, ggplot2, and pheatmap.
library(DESeq2)
library(ggplot2)
library(pheatmap)
Reading in files and basic data exploration.
First, before we read anything into R, what is the full path to the raw counts file?
Solution
/data/RNA/bulk/airway_raw_counts.csv.gz
Use this to read into R using the read.csv command.
Let’s read the help message for this command.
?read.csv
Main argument here is file. Let’s try filling in the path from above.
Solution
read.csv(file=/data/RNA/bulk/airway_raw_counts.csv.gz)Error in parse(text = x, srcfile = src): <text>:1:15: unexpected '/' 1: read.csv(file=/ ^Traceback:
Oops, let’s fix that.
Solution
read.csv(file="/data/RNA/bulk/airway_raw_counts.csv.gz")A data.frame: 63677 × 9 gene_id SRR1039508 SRR1039509 SRR1039512 SRR1039513 SRR1039516 SRR1039517 SRR1039520 SRR1039521 <chr> <int> <int> <int> <int> <int> <int> <int> <int> ENSG00000000003 679 448 873 408 1138 1047 770 572 ENSG00000000005 0 0 0 0 0 0 0 0 ENSG00000000419 467 515 621 365 587 799 417 508 ENSG00000000457 260 211 263 164 245 331 233 229 ENSG00000000460 60 55 40 35 78 63 76 60 ENSG00000000938 0 0 2 0 1 0 0 0 ENSG00000000971 3251 3679 6177 4252 6721 11027 5176 7995 ENSG00000001036 1433 1062 1733 881 1424 1439 1359 1109 ENSG00000001084 519 380 595 493 820 714 696 704 ENSG00000001167 394 236 464 175 658 584 360 269 ... ENSG00000273484 0 0 0 0 0 0 0 0 ENSG00000273485 2 3 1 1 1 1 1 0 ENSG00000273486 14 11 25 8 20 32 12 11 ENSG00000273487 5 9 4 11 10 10 4 10 ENSG00000273488 7 5 8 3 8 16 11 14 ENSG00000273489 0 0 0 1 0 1 0 0 ENSG00000273490 0 0 0 0 0 0 0 0 ENSG00000273491 0 0 0 0 0 0 0 0 ENSG00000273492 0 0 1 0 0 0 0 0 ENSG00000273493 0 0 0 0 1 0 0 0
Looks like we just read in as standard input/output, rather than saving to an object.
Let’s save the result of this command in an object called raw.counts.
Solution
raw.counts = read.csv(file="/data/RNA/bulk/airway_raw_counts.csv.gz")
Use the head command to look at the first few lines.
head(raw.counts)
gene_id SRR1039508 SRR1039509 SRR1039512 SRR1039513 SRR1039516
1 ENSG00000000003 679 448 873 408 1138
2 ENSG00000000005 0 0 0 0 0
3 ENSG00000000419 467 515 621 365 587
4 ENSG00000000457 260 211 263 164 245
5 ENSG00000000460 60 55 40 35 78
6 ENSG00000000938 0 0 2 0 1
SRR1039517 SRR1039520 SRR1039521
1 1047 770 572
2 0 0 0
3 799 417 508
4 331 233 229
5 63 76 60
6 0 0 0
Looks like the first column is the gene ids.
It would be better if we could read in such that it would automatically make those the row names, so that all the values in the table could be numeric.
Let’s look at the read.csv help message again to see if there is a way to do that.
?read.csv
If we scroll down we find the following:
row.names: a vector of row names. This can be a vector giving the
actual row names, or a single number giving the column of the
table which contains the row names, or character string
giving the name of the table column containing the row names.
Let’s use this argument to read in the file again using read.csv, this time taking the first column as the row names.
Solution
raw.counts = read.csv(file="/data/RNA/bulk/airway_raw_counts.csv.gz",row.names=1)
Look at the first few lines of raw.counts again.
head(raw.counts)
SRR1039508 SRR1039509 SRR1039512 SRR1039513 SRR1039516
ENSG00000000003 679 448 873 408 1138
ENSG00000000005 0 0 0 0 0
ENSG00000000419 467 515 621 365 587
ENSG00000000457 260 211 263 164 245
ENSG00000000460 60 55 40 35 78
ENSG00000000938 0 0 2 0 1
SRR1039517 SRR1039520 SRR1039521
ENSG00000000003 1047 770 572
ENSG00000000005 0 0 0
ENSG00000000419 799 417 508
ENSG00000000457 331 233 229
ENSG00000000460 63 76 60
ENSG00000000938 0 0 0
Looks good! Think we are ready to proceed with this object now.
Check how many rows there are.
nrow(raw.counts)
[1] 63677
So, we have 63,677 genes, and 8 samples (as we saw when we did head).
Next, read in the file with the study design.
Remember path to this file is:
/data/RNA/bulk/airway_sample_metadata.csv
We can once again use the csv command, and take the first column as row names.
Read into an object called expdesign.
Solution
expdesign = read.csv(file="/data/RNA/bulk/airway_sample_metadata.csv",row.names=1)
Look at the object.
expdesign
cell dex avgLength
SRR1039508 N61311 untrt 126
SRR1039509 N61311 trt 126
SRR1039512 N052611 untrt 126
SRR1039513 N052611 trt 87
SRR1039516 N080611 untrt 120
SRR1039517 N080611 trt 126
SRR1039520 N061011 untrt 101
SRR1039521 N061011 trt 98
We are mainly interested in columns cell (says which of the four cell lines the sample is) and dex (says whether or not the sample had drug treatment) here.
Making a DESeq object using DESeq2
We are going to take the raw counts matrix, and the experimental design matrix, and make a special type of object called a DESeqDataSet.
We are going to use a function DESeqDataSetFromMatrix, from the DESeq2 library that we already loaded, to do this.
Let’s view the help message for that function.
?DESeqDataSetFromMatrix
Start of the help message looks like this:
DESeqDataSetFromMatrix(
countData,
colData,
design,
tidy = FALSE,
ignoreRank = FALSE,
...
)
We also find the following if we scroll down to the example:
countData <- matrix(1:100,ncol=4)
condition <- factor(c("A","A","B","B"))
dds <- DESeqDataSetFromMatrix(countData, DataFrame(condition), ~ condition)
If we explicitly stated the arguments in the example (and named the objects less confusingly), it would look like this:
mycounts = matrix(1:100,ncol=4)
group = factor(c("A","A","B","B"))
expdesign_example = data.frame(condition = group)
#expdesign_example now looks like this:
# condition
#1 A
#2 A
#3 B
#4 B
dds = DESeqDataSetFromMatrix(countData = mycounts,
colData = expdesign_example,
design = ~condition)
Let’s run this for our data set, but using raw.counts as the count matrix and expdesign as the design matrix.
For the design, let’s look at the column names of expdesign again.
colnames(expdesign)
[1] "cell" "dex" "avgLength"
Here, we want to include both the cell line (cell) and treatment status (dex) in the design.
Save to an object called myDESeqObject.
Solution
myDESeqObject = DESeqDataSetFromMatrix(countData = raw.counts, colData = expdesign, design = cell,dex)Error: object 'dex' not found
Oops, looks like we did something wrong.
Should we be putting cell and dex in quotes maybe? That worked the last time we got this kind of error.
Solution
myDESeqObject = DESeqDataSetFromMatrix(countData = raw.counts, colData = expdesign, design = c("cell","dex"))Error in DESeqDataSet(se, design = design, ignoreRank) : 'design' should be a formula or a matrix
Hm, still not right.
I think what we are looking for is a design formula here.
If you search for how to make a design formula, you can find this guide, where we seem to see the following rules:
- Design formula should always start with
~. - If more than one variable, put a
+in between. - Nothing in quotes.
For example, their formula for the variables diet and sex was this:
~diet + sex
Let’s see if we can finally fix our command to properly set the design to include cell and dex here.
Solution
myDESeqObject = DESeqDataSetFromMatrix(countData = raw.counts, colData = expdesign, design = ~cell + dex)
When we run the above, we get the following output:
Warning message:
In DESeqDataSet(se, design = design, ignoreRank) :
some variables in design formula are characters, converting to factors
This is just a warning message, doesn’t mean we did anything wrong.
Looks like the command finally ran OK!
Normalization on the DESeq object
We will next run the estimateSizeFactors command on this object, and save the output back to myDESeqObject.
This command prepares the object for the next step, where we normalize the data by library size (total counts).
myDESeqObject = estimateSizeFactors(myDESeqObject)
Next, we will run a normalization method called rlogTransformation (regularized-log transformation) on myDESeqObject, and save to a new object called rlogObject.
Basic syntax for how this works:
transformedObject = transformationCommand(object = oldObject)
Replace transformedObject here with the name we want for the new object, and transformationCommand with rlogTransformation. Here the oldObject is the DESeq object we already made.
Solution
rlogObject = rlogTransformation(object = myDESeqObject)
Let’s also save just the normalized values from rlogObject in a separate object called rlogMatrix.
rlogMatrix = assay(rlogObject)
Principal components analysis (PCA)
We will run DESeq2’s plotPCA function on the object produced by the rlogTransformation command, to create a plot where we can see the relationship between samples by reducing into two dimensions.
Again, let’s view the help message for this function.
?plotPCA
We find the following documentation.
Usage:
## S4 method for signature 'DESeqTransform'
plotPCA(object, intgroup = "condition", ntop = 500, returnData = FALSE)
Arguments:
object: a ‘DESeqTransform’ object, with data in ‘assay(x)’, produced
for example by either ‘rlog’ or
‘varianceStabilizingTransformation’.
intgroup: interesting groups: a character vector of names in
‘colData(x)’ to use for grouping
Examples:
dds <- makeExampleDESeqDataSet(betaSD=1)
vsd <- vst(dds, nsub=500)
plotPCA(vsd)
A more explicitly stated version of the example:
dds = makeExampleDESeqDataSet(betaSD=1)
raw.counts_example = assay(dds)
expdesign_example = colData(dds)
head(raw.counts_example)
expdesign_example
sample1 sample2 sample3 sample4 sample5 sample6 sample7 sample8 sample9 sample10 sample11 sample12
gene1 6 6 2 8 17 2 0 4 17 7 3 10
gene2 27 48 40 26 12 39 7 5 4 14 9 6
gene3 8 7 4 2 1 1 2 21 4 2 9 0
gene4 36 24 41 25 44 25 39 19 8 31 17 62
gene5 22 36 10 28 33 9 55 58 71 12 68 22
gene6 1 12 5 6 15 1 2 0 7 4 0 1
DataFrame with 12 rows and 1 column
condition
<factor>
sample1 A
sample2 A
sample3 A
sample4 A
sample5 A
... ...
sample8 B
sample9 B
sample10 B
sample11 B
sample12 B
dds = DESeqDataSetFromMatrix(countData = raw.counts_example,
colData = expdesign_example,
design=~condition)
vsd <- vst(dds, nsub=500)
plotPCA(object = vsd)
We already ran DESeqDataSetFromMatrix, as well as transformation (we ran rlogTransformation instead of vst, but same idea).
So, just need to plug the object from rlogTransformation into plotPCA.
Solution
plotPCA(object = rlogObject)Error in .local(object, ...) : the argument 'intgroup' should specify columns of colData(dds)
In the example, we did not need to explicitly state what variable to color by in the plot, since there was only one (condition).
However here, we have two possible variables , so we need to explicitly state.
In the example, if we had explicitly stated to color by condition, it would look like this.
plotPCA(object = vsd, intgroup = "condition")
Let’s use the same syntax here to try running plotPCA on rlogObject again, this time explicitly stating to color by cell using the intgroup argument.
Solution
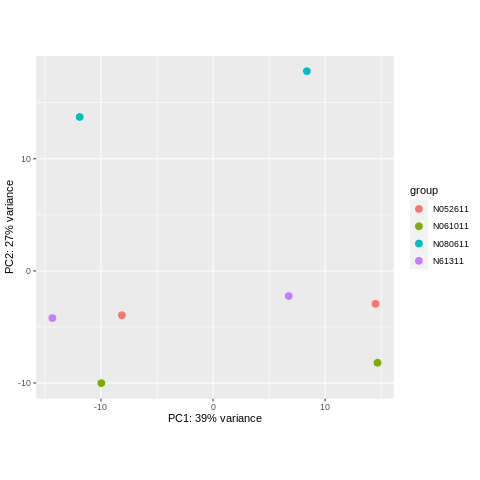
plotPCA(object = rlogObject, intgroup = "cell")
Next, repeat but this time color by dex.
Solution
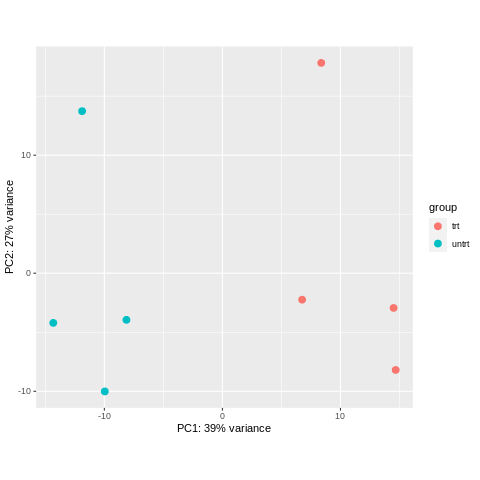
plotPCA(object = rlogObject, intgroup = "dex")
Interpreting the PCA plot
Let’s look at the two plots we made, and answer the following.
- What principal component separates the four different cell lines? Which one separates treated vs. untreated?
- How much variance is explained by each principal component (hint: see axis labels)?
Solution
PC2 separates the four different cell lines. PC1 separates treated vs. untreated.
PC1 explains 39% of the variance. PC2 explains 27% of the variance.
Differential expression testing
Now that we have created the DESeq object, and done some initial data exploration, it is time to run differential expression testing to see which genes are significantly different between conditions (here, drug-treated vs. not drug-treated).
One step we need to run to get these results is the command DESeq. Let’s pull up the help message for this.
?DESeq
Start of this help message:
Usage:
DESeq(
object,
test = c("Wald", "LRT"),
fitType = c("parametric", "local", "mean", "glmGamPoi"),
sfType = c("ratio", "poscounts", "iterate"),
betaPrior,
full = design(object),
reduced,
quiet = FALSE,
minReplicatesForReplace = 7,
modelMatrixType,
useT = FALSE,
minmu = if (fitType == "glmGamPoi") 1e-06 else 0.5,
parallel = FALSE,
BPPARAM = bpparam()
)
Arguments:
object: a DESeqDataSet object, see the constructor functions
‘DESeqDataSet’, ‘DESeqDataSetFromMatrix’,
‘DESeqDataSetFromHTSeqCount’.
Seems like we can just input our DESeqDataSet object to the object argument?
Let’s do that, and save as a new object called dds.
dds = DESeq(object = myDESeqObject)
If all goes well, you should get the following output as the command runs.
using pre-existing size factors
estimating dispersions
gene-wise dispersion estimates
mean-dispersion relationship
final dispersion estimates
fitting model and testing
Next, we will need to run the results function on dds.
Now, pull up the help message for that function.
?results
Usage:
results(
object,
contrast,
name,
lfcThreshold = 0,
...)
Arguments:
object: a DESeqDataSet, on which one of the following functions has
already been called: ‘DESeq’, ‘nbinomWaldTest’, or
‘nbinomLRT’
contrast: this argument specifies what comparison to extract from the
‘object’ to build a results table. one of either:
• a character vector with exactly three elements: the name
of a factor in the design formula, the name of the
numerator level for the fold change, and the name of the
denominator level for the fold change (simplest case)
...
Examples:
## Example 1: two-group comparison
dds <- makeExampleDESeqDataSet(m=4)
dds <- DESeq(dds)
res <- results(dds, contrast=c("condition","B","A"))
To highlight the example:
dds <- DESeq(dds)
res <- results(dds, contrast=c("condition","B","A"))
Replace condition, B, and A with the appropriate values for this data set.
We are looking to compare the effect of treatment with the drug.
Let’s look at our experimental design matrix again.
expdesign
cell dex avgLength
SRR1039508 N61311 untrt 126
SRR1039509 N61311 trt 126
SRR1039512 N052611 untrt 126
SRR1039513 N052611 trt 87
SRR1039516 N080611 untrt 120
SRR1039517 N080611 trt 126
SRR1039520 N061011 untrt 101
SRR1039521 N061011 trt 98
Filling in the contrast argument to
resultsfrom expdesignSome questions we need to answer here are:
- What is the variable name for drug treatment here?
- What are the two levels of this variable?
- What order should we put these two levels in, if we want untreated to be the baseline?
Solution
- The variable name for drug treatment is
dex.- The two levels are
trtanduntrt.- We should put
trtfirst anduntrtsecond so that the fold-change will be expressed as treated/untreated.
Now that we figured that out, we just need to plug everything into the function.
Output to an object called res.
Solution
res = results(object = dds, contrast = c("dex","trt","untrt"))
Let’s look at the first few rows of the results using head.
head(res)
log2 fold change (MLE): dex trt vs untrt
Wald test p-value: dex trt vs untrt
DataFrame with 6 rows and 6 columns
baseMean log2FoldChange lfcSE stat pvalue
<numeric> <numeric> <numeric> <numeric> <numeric>
ENSG00000000003 708.602170 -0.3812540 0.100654 -3.787752 0.000152016
ENSG00000000005 0.000000 NA NA NA NA
ENSG00000000419 520.297901 0.2068126 0.112219 1.842943 0.065337292
ENSG00000000457 237.163037 0.0379204 0.143445 0.264356 0.791505742
ENSG00000000460 57.932633 -0.0881682 0.287142 -0.307054 0.758801924
ENSG00000000938 0.318098 -1.3782270 3.499873 -0.393793 0.693733530
padj
<numeric>
ENSG00000000003 0.00128292
ENSG00000000005 NA
ENSG00000000419 0.19646985
ENSG00000000457 0.91141962
ENSG00000000460 0.89500478
ENSG00000000938 NA
Run the following to get an explanation of what each column in this output means.
mcols(res)$description
[1] "mean of normalized counts for all samples"
[2] "log2 fold change (MLE): dex trt vs untrt"
[3] "standard error: dex trt vs untrt"
[4] "Wald statistic: dex trt vs untrt"
[5] "Wald test p-value: dex trt vs untrt"
[6] "BH adjusted p-values"
Ready to start to explore and interpret these results.
Visualizing expression of differentially expressed genes (heatmap)
Use the which function to get the indices (row numbers) of the genes with adjusted p-value < .01 (false discovery rate of 1%).
Then, save as an object called degs.
First, which column index or name contains the adjusted p-values?
Solution
Column "padj", or column number 6.
Then, we will need to extract this column. A few possible ways listed below.
Solution
res$padj res[,6] res[,"padj"]
Next, help message for the which command (from base package) gives the following:
Usage:
which(x, arr.ind = FALSE, useNames = TRUE)
arrayInd(ind, .dim, .dimnames = NULL, useNames = FALSE)
Arguments:
x: a ‘logical’ vector or array. ‘NA’s are allowed and omitted
(treated as if ‘FALSE’).
Examples:
which(LETTERS == "R")
..
which(1:10 > 3, arr.ind = TRUE)
So, we just need to add a statement to get only values less than 0.01 from the column.
Solution
degs = which(res$padj < .01)
How many differentially expressed genes do we find?
length(degs)
[1] 2901
Next, let’s subset the normalized expression matrix (rlogMatrix) to only these genes, and output to a new matrix rlogMatrix.degs.
Syntax of matrix subsetting:
mydat.subset = mydat[indices_to_subset,]
Solution
rlogMatrix.degs = rlogMatrix[degs,]
Let’s run the pheatmap command on this matrix to output a heatmap to the console.
pheatmap(object = rlogMatrix.degs)
Error in pheatmap(object = rlogMatrix.degs) :
argument "mat" is missing, with no default
Well, that’s why we should read the help message! Seems the main argument for the input object to pheatmap is mat, not object.
pheatmap(mat = rlogMatrix.degs)
Does not seem super informative!
Let’s look at the pheatmap help message again and see if we can add some arguments to make it better.
?pheatmap
Scrolling down to arguments, we see:
scale: character indicating if the values should be centered and
scaled in either the row direction or the column direction,
or none. Corresponding values are ‘"row"’, ‘"column"’ and
‘"none"’
Let’s scale across the genes, so by row.
And also this:
show_rownames: boolean specifying if row names are be shown.
Boolean means either TRUE or FALSE. Let’s turn this off.
Solution
pheatmap(mat = rlogMatrix.degs, scale="row", show_rownames=FALSE)
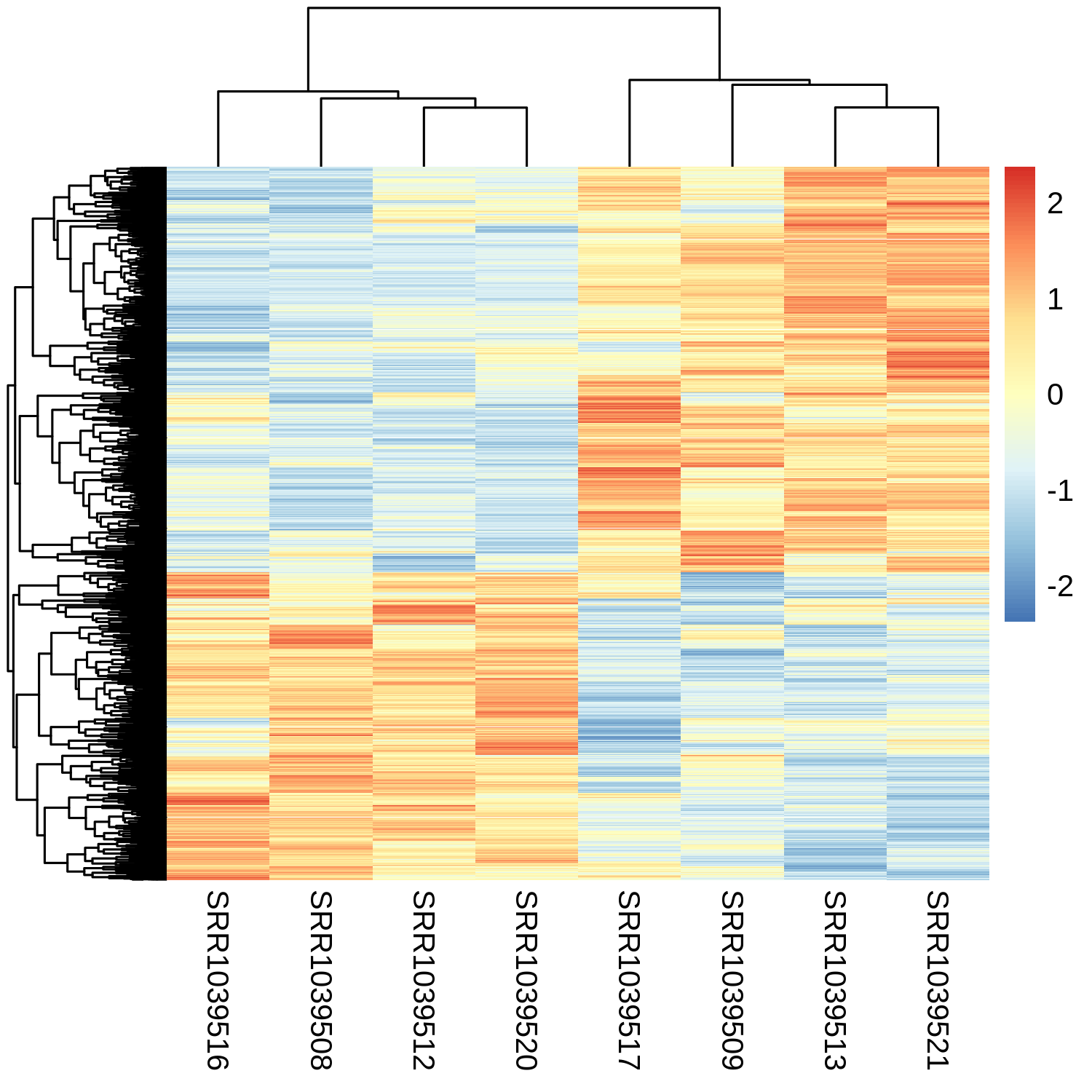
Think we mostly have what we want now.
Except, the “SRR” sample names are not very informative.
It would be good to have an annotation bar at the top of the heatmap that said what cell line each sample is from, and whether it is treated or untreated.
Going back to the pheatmap help message:
annotation_row: data frame that specifies the annotations shown on left
side of the heatmap. Each row defines the features for a
specific row. The rows in the data and in the annotation are
matched using corresponding row names. Note that color
schemes takes into account if variable is continuous or
discrete.
annotation_col: similar to annotation_row, but for columns.
We already have a data frame with the annotation we are looking for, so just need to give it as an argument here.
Solution
pheatmap(mat = rlogMatrix.degs, scale="row", show_rownames=FALSE, annotation_row=expdesign)Error in seq.int(rx[1L], rx[2L], length.out = nb) : 'from' must be a finite number In addition: Warning messages: 1: In min(x) : no non-missing arguments to min; returning Inf 2: In max(x) : no non-missing arguments to max; returning -Inf
Oops - looks like we used the wrong argument. We want to annotate the samples, not the genes.
Solution
pheatmap(mat = rlogMatrix.degs, scale="row", show_rownames=FALSE, annotation_col=expdesign)
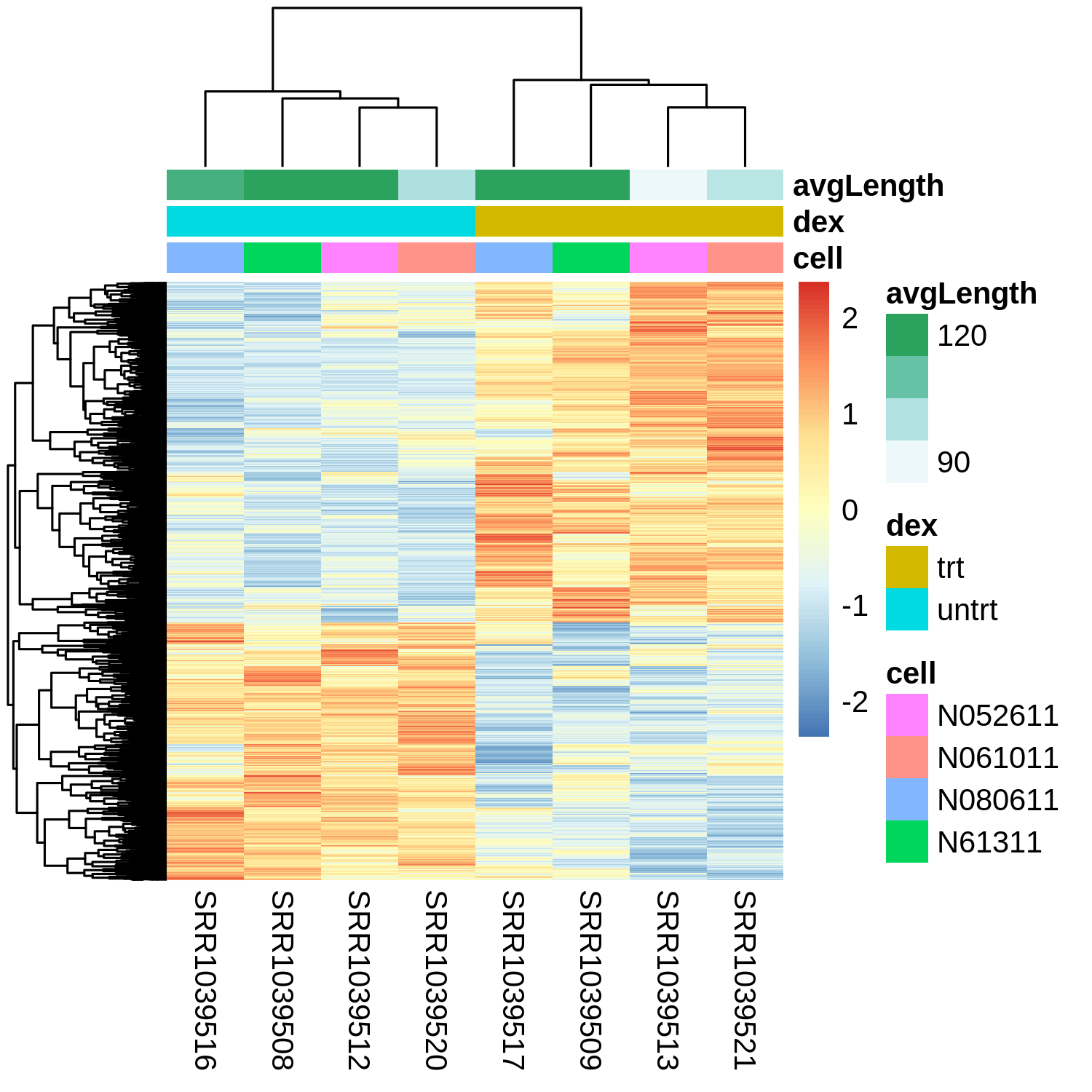
Looks like the samples separate primarily by treatment, as we would expect here since these are the differentially expressed genes.
We also see some variability by cell line, as we would have expected from the PCA plot.
Finally, we see a mix of genes upregulated vs. downregulated in the treated condition.
Visualizing differential expression statistics (scatterplots)
MA plot
In the above section, we only looked at the differentially expressed genes.
However, we may also want to look at patterns in the differential expression results overall, including genes that did not reach significance.
One common summary plot from differential expression is the MA plot.
In this plot, mean expression across samples (baseMean) is compared to log-fold-change, with genes colored by whether or not they meet the significance level we set for adjusted p-value (padj).
Generally for lowly expressed genes, the difference between conditions has to be stronger to create statistically significant results compared to highly expressed genes. The MA plot shows this effect.
DESeq2 has a nice function plotMA that will do this for you (using the output of the results function), and format everything nicely.
Add argument alpha=0.01 to set the adjusted p-value cutoff to 0.01 instead of the default 0.1.
plotMA(object = res,alpha=0.01)
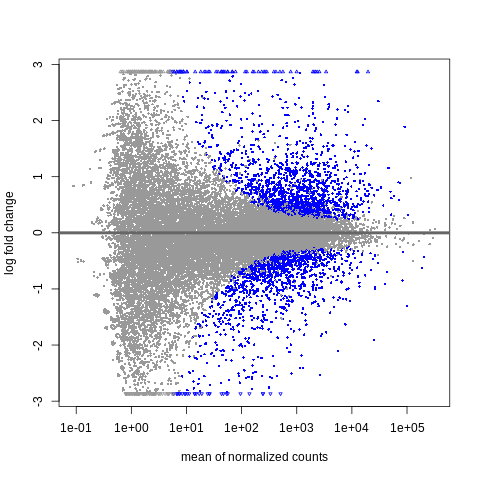
The x-axis here is log10-scaled. The y-axis here is log2(treated/untreated).
The blue dots are for significantly differentially expressed genes (padj < .01).
We find that at low baseMean (closer to 1e+01 or ~10), the magnitude of the log2-fold-change must be very large (often +/-2 or +/- 3, so something like 4-fold or 8-fold difference) for the gene to be significant.
Meanwhile at higher baseMean (say, as we go to 1e+03 or ~1000 reads and above), we find that the magnitude of the log2-fold-change can be much smaller (going down to +/- 0.5, so something like a 1.4-fold difference, or even less).
Volcano plot
Another interesting plot is a volcano plot. This is a plot showing the relationship between the log2-fold-change and the adjusted p-value.
First, extract these two values from res.
Let’s refresh ourselves on what the column names are here.
colnames(res)
[1] "baseMean" "log2FoldChange" "lfcSE" "stat"
[5] "pvalue" "padj"
Extract columns log2FoldChange and padj into objects of the same name.
Solution
log2FoldChange = res$log2FoldChange padj = res$padj
Plot log2FoldChange on the x-axis and padj on the y-axis.
Solution
plot(log2FoldChange,padj)
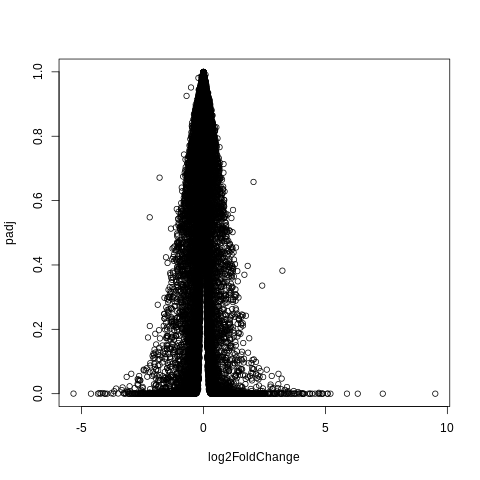
Add argument to make the points smaller.
Argument:
pch="."
Solution
plot(log2FoldChange,padj,pch=".")
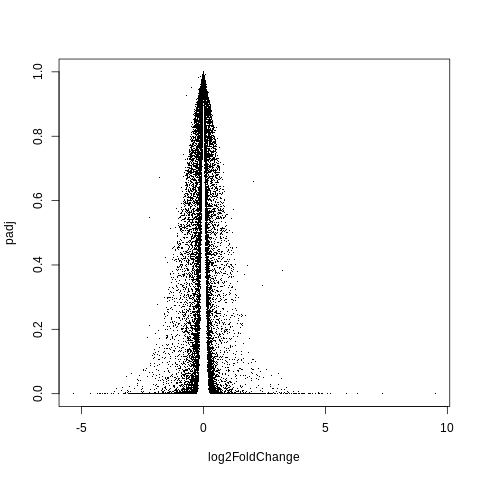
Hm, this still doesn’t look quite right.
If I search for what a volcano plot should look like, I get something closer to this.
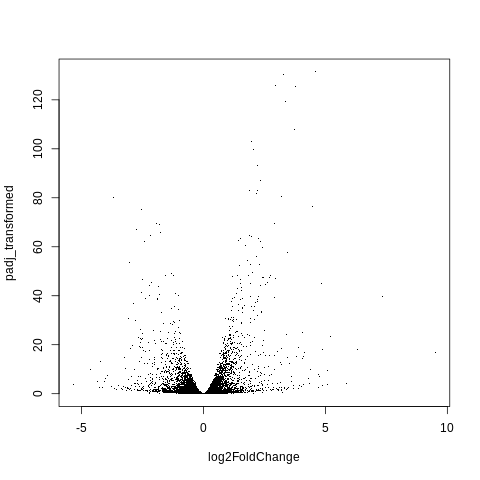
I believe we need to take -log10 of padj.
Let’s do that, and save as padj_transformed.
Then, redo plot using this new variable.
Solution
padj_transformed = -log10(padj) plot(log2FoldChange,padj_transformed,pch=".")
Preparing files for functional enrichment (ORA/GSEA)
Next, we will want to take the results of differential expression testing, and prepare for input into functional enrichment, either over-representation (ORA) or gene set enrichment (GSEA) analysis.
Then, we will import these inputs to a web-based tool called WebGestalt.
In over-representation analysis, the idea is to test whether genes that are significantly differential expressed are enriched for certain categories of genes (gene sets). The input to this is a list of the gene IDs/names for the differentially expressed genes.
In gene set enrichment analysis, genes are ranked by how differential expressed they are, and whether they are upregulated or downregulated. The input to this is a table with the gene IDs/names of all genes, and the test-statistic (here the stat column), ordered by the test-statistic.
Preparing for input into ORA
Here, we will prepare the input for ORA by first getting all the gene IDs into an object called geneids.
These gene IDs are stored in the row names of the results (res), so we can get them out using the rownames function.
Solution
geneids = rownames(res)
Next, subset geneids to just the differentially expressed genes using the previously calculated indices (that we made for the heatmap step).
Syntax to subset an object using previously calculated indices.
x_subset = x[previously_calculated_indices]
Let’s output to an object called geneids.sig.
Solution
geneids.sig = geneids[degs]
Output to a text file called geneids_sig.txt using the writeLines command.
Let’s get the syntax for this command.
?writeLines
Usage:
writeLines(text, con = stdout(), sep = "\n", useBytes = FALSE)
Arguments:
text: A character vector
con: A connection object or a character string.
There is no example in the help message, but I found one on Stack Overflow that may be helpful (paraphrased below).
mywords = c("Hello","World")
output_file = "output.txt"
writeLines(text = mywords, con = output_file)
Let’s run this here.
Solution
output_file = geneids_sig.txt writeLines(text = geneids.sig, con = output_file)Error: object 'geneids_sig.txt' not found
Oops, let’s fix.
Solution
output_file = "geneids_sig.txt" writeLines(text = geneids.sig, con = output_file)
Go to the Terminal tab, and let’s look at this file in more detail.
First, do head and tail.
head geneids_sig.txt
ENSG00000000003
ENSG00000000971
ENSG00000001167
ENSG00000002834
ENSG00000003096
ENSG00000003402
ENSG00000004059
ENSG00000004487
ENSG00000004700
ENSG00000004799
tail geneids_sig.txt
ENSG00000272695
ENSG00000272761
ENSG00000272796
ENSG00000272841
ENSG00000272870
ENSG00000273038
ENSG00000273131
ENSG00000273179
ENSG00000273259
ENSG00000273290
Also check number of lines.
wc -l geneids_sig.txt
2901 geneids_sig.txt
All looks good.
Normally, you would download the file you just created to your laptop, but we may not always have a way to transfer files from the cloud VM to your laptop. So, let’s download the file from the Github for this workshop. Download link here.
Preparing for input into GSEA
Let’s move on to creating the input for GSEA. This will be a data frame with two columns.
gene: The gene ID (already have these for all genes ingeneids)stat: Thestatcolumn from the differential expression results (res)
We will use the data.frame function here.
?data.frame
Examples:
L3 <- LETTERS[1:3]
char <- sample(L3, 10, replace = TRUE)
d <- data.frame(x = 1, y = 1:10, char = char)
Let’s call this data frame gsea.input.
Solution
gsea.input = data.frame(gene = geneids, stat = res$stat)
Sort by the stat column.
Solution
order_genes = order(gsea.input$stat) gsea.input = gsea.input[order_genes,]
Output to a tab-delimited text file gsea_input.txt using the write.table function.
Solution
output_file = "gsea_input.txt" write.table(x = gsea.input,file=output_file,sep="\t")
Let’s switch over to the Terminal tab and look at this file in more detail.
head gsea_input.txt
"gene" "stat"
"10923" "ENSG00000162692" -19.4160587914648
"14737" "ENSG00000178695" -18.8066074036207
"3453" "ENSG00000107562" -18.0775817519565
"9240" "ENSG00000148848" -18.0580936880167
"8911" "ENSG00000146250" -17.7950173361052
"3402" "ENSG00000106976" -17.6137734750388
"11121" "ENSG00000163394" -17.4484303669187
"5627" "ENSG00000124766" -17.1299063626842
"3259" "ENSG00000105989" -15.8949963870475
Info on formatting of the file for input into GSEA available here, from the Broad Institute’s documentation.
This says that there should be two columns - one for the feature identifiers (i.e. gene IDs or symbols) and one for the weight of the gene (here, the stat column). But our output has three columns - this is not what we want.
Let’s go back to the R notebook tab and see if we can fix this.
Comparing to the example in the file format documentation, it seems we should do the following:
- Remove the quotation marks.
- Remove the header.
- Remove the first column.
Go back to R and see if we can find arguments in the read.table function to help with this.
?read.table
Arguments:
quote: a logical value (‘TRUE’ or ‘FALSE’) or a numeric vector. If
‘TRUE’, any character or factor columns will be surrounded by
double quotes. If a numeric vector, its elements are taken
as the indices of columns to quote. In both cases, row and
column names are quoted if they are written. If ‘FALSE’,
nothing is quoted.
row.names: either a logical value indicating whether the row names of
‘x’ are to be written along with ‘x’, or a character vector
of row names to be written.
col.names: either a logical value indicating whether the column names
of ‘x’ are to be written along with ‘x’, or a character
vector of column names to be written. See the section on
‘CSV files’ for the meaning of ‘col.names = NA’.
Looks like we need to add a few arguments to get our output formatted correctly.
Let’s output to a new file gsea_input_corrected.txt.
Solution
output_file = "gsea_input_corrected.txt" write.table(x = gsea.input,file=output_file,sep="\t",row.names=FALSE,col.names=FALSE,quote=FALSE)
Go back to the Terminal tab, and look at this file again.
head gsea_input_corrected.txt
ENSG00000162692 -19.4160587914648
ENSG00000178695 -18.8066074036207
ENSG00000107562 -18.0775817519565
ENSG00000148848 -18.0580936880167
ENSG00000146250 -17.7950173361052
ENSG00000106976 -17.6137734750388
ENSG00000163394 -17.4484303669187
ENSG00000124766 -17.1299063626842
ENSG00000105989 -15.8949963870475
ENSG00000108821 -15.2668170602372
Looks ok - what about tail?
tail gsea_input_corrected.txt
ENSG00000273470 NA
ENSG00000273471 NA
ENSG00000273475 NA
ENSG00000273479 NA
ENSG00000273480 NA
ENSG00000273481 NA
ENSG00000273482 NA
ENSG00000273484 NA
ENSG00000273490 NA
ENSG00000273491 NA
Let’s also check the line count.
wc -l gsea_input_corrected.txt
63677 gsea_input_corrected.txt
Hm - don’t think we should be including genes with an NA for the stat column?
Let’s fix again.
Let’s use grep to remove lines with “NA”, and output to a new file gsea_input_corrected_minus_NA.txt.
Solution
grep -v NA gsea_input_corrected.txt > gsea_input_corrected_minus_NA.txt
Check again.
head gsea_input_corrected_minus_NA.txt
ENSG00000162692 -19.4160587914648
ENSG00000178695 -18.8066074036207
ENSG00000107562 -18.0775817519565
ENSG00000148848 -18.0580936880167
ENSG00000146250 -17.7950173361052
ENSG00000106976 -17.6137734750388
ENSG00000163394 -17.4484303669187
ENSG00000124766 -17.1299063626842
ENSG00000105989 -15.8949963870475
ENSG00000108821 -15.2668170602372
tail gsea_input_corrected_minus_NA.txt
ENSG00000154734 20.2542354089516
ENSG00000125148 20.9292144890544
ENSG00000162614 21.6140905458227
ENSG00000157214 21.968453609644
ENSG00000211445 22.4952633774851
ENSG00000189221 23.6530144138538
ENSG00000101347 24.2347153246759
ENSG00000120129 24.2742584224751
ENSG00000165995 24.7125510867281
ENSG00000152583 24.8561114516005
wc -l gsea_input_corrected_minus_NA.txt
33469 gsea_input_corrected_minus_NA.txt
We now have fewer lines, because we removed the lines for the genes with an “NA” in the stat column.
You can download a copy of this file here.
Running functional enrichment (ORA/GSEA)
Let’s head to the website WebGestalt.
Run ORA
Click “Click to upload” next to “Upload ID List”.
Upload geneids_sig.txt.
For the reference set, select “genome protein-coding”.
In advanced parameters, switch from top 10 to FDR 0.05.
Let’s look at the results!
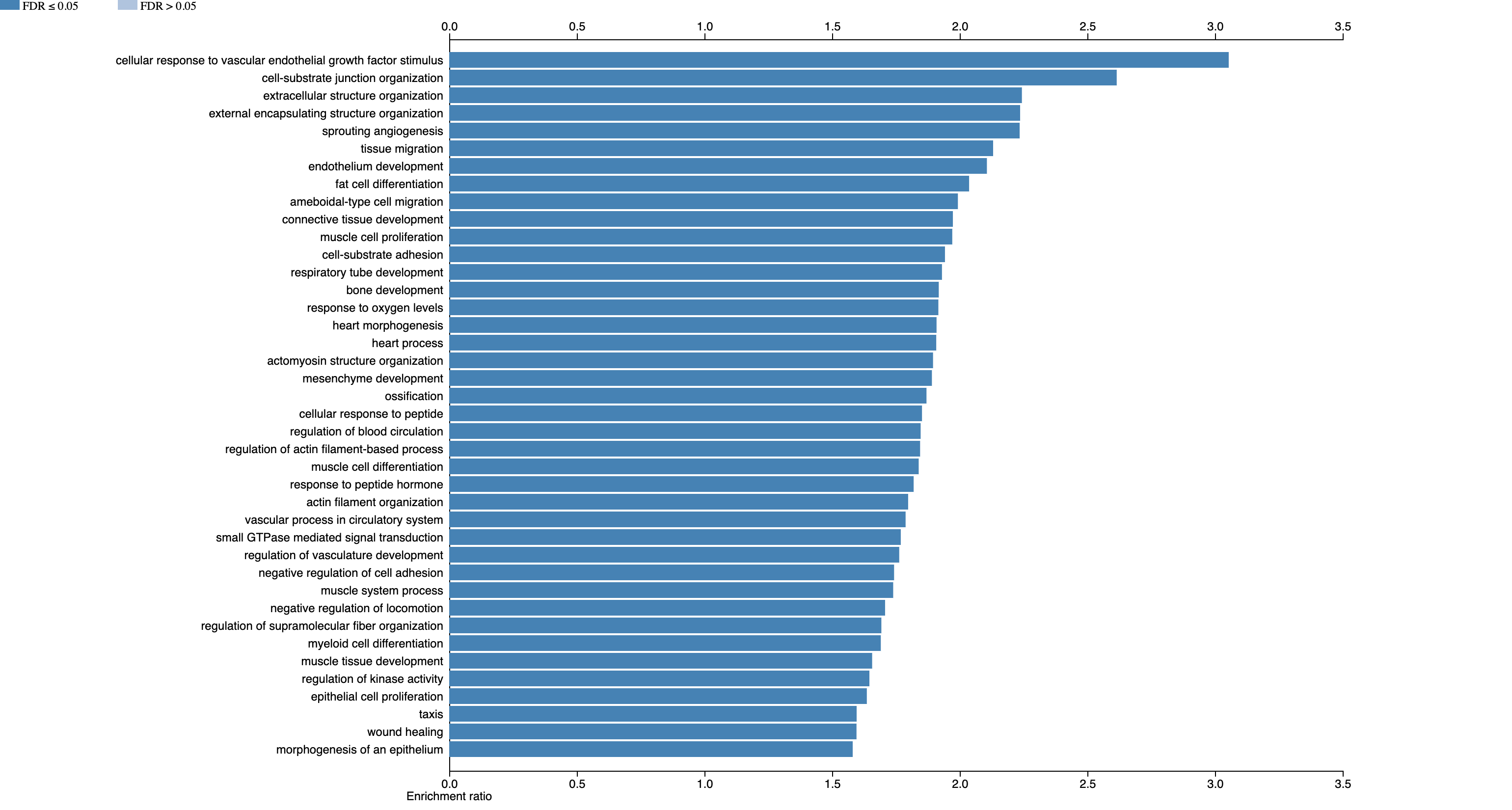
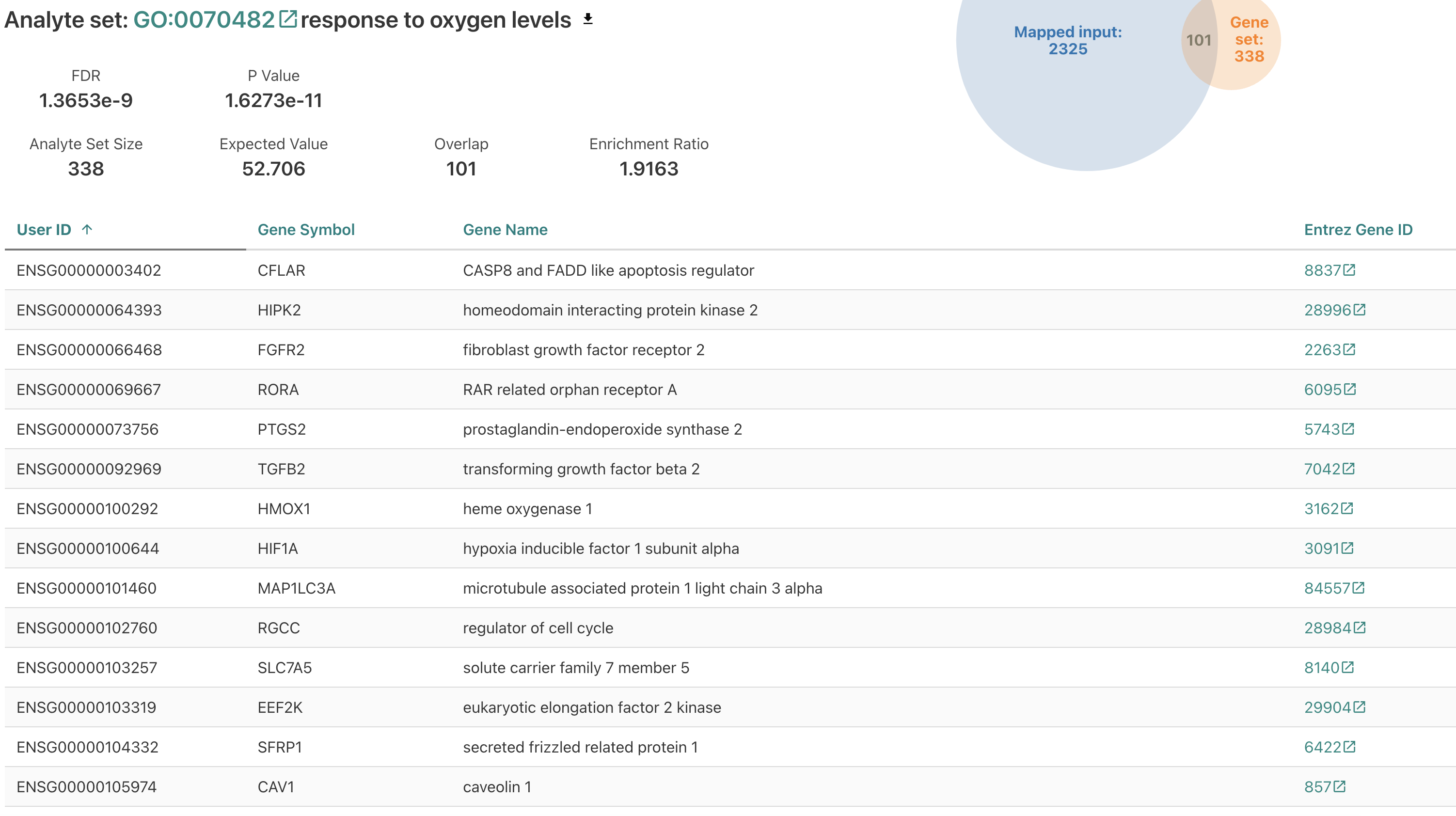
Let’s scroll down and click on “response to oxygen levels”, as this seems pretty relevant for an airway dataset.
We can scroll through and look at the genes that are in this gene set, that are differentially expressed.
Run GSEA.
Let’s move on to running gene set enrichment analysis (GSEA).
For the tab at the top, switch to “Gene Set Enrichment Analysis”.
Switch back the functional database to the same as before (geneontology, Biological Process noRedundant).
This time, upload gsea_input_corrected_minus_NA.txt.
Also reset the advanced parameters to select based on FDR of 0.05 rather than the top 10 again.
We get the following error message.

Turns out, this is as simple as renaming the file!
Let’s change the file suffix to “.rnk” instead of “.txt” (rename the file).
Below is shown how to do this using Mac’s Finder program - you can do this however you would normally for your system.
Now, go back and redo all of the above, but input gsea_input_corrected_minus_NA.rnk.
This time it should work, and output something like the following:
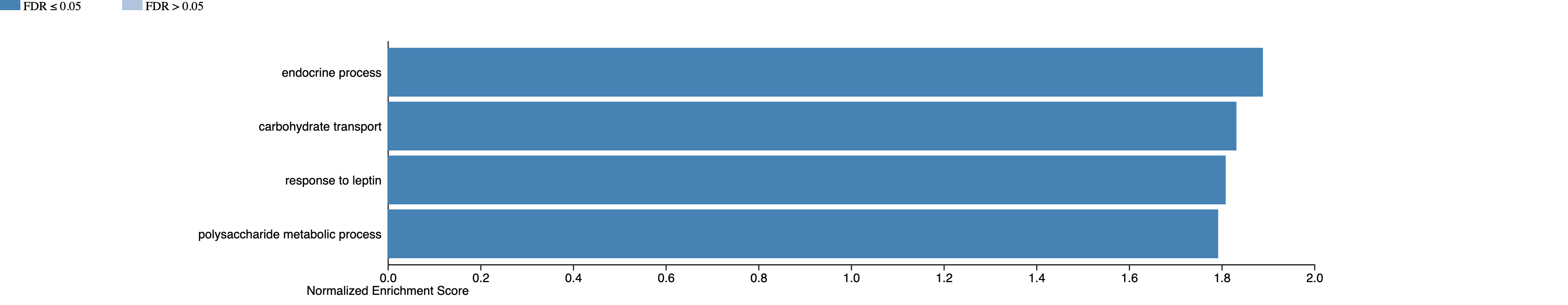
Hm - fewer categories than we might have expected?
Let’s redo, but change functional database to pathway = Kegg.
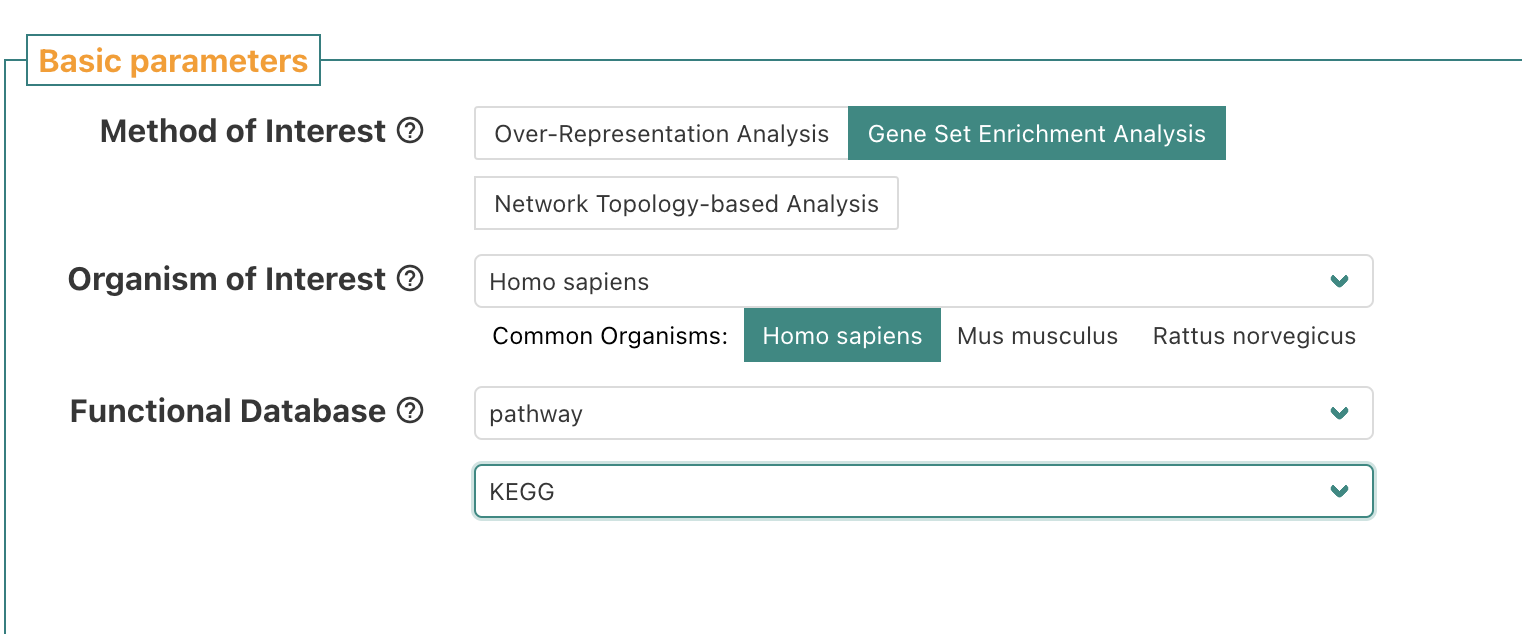

Click on autophagy.
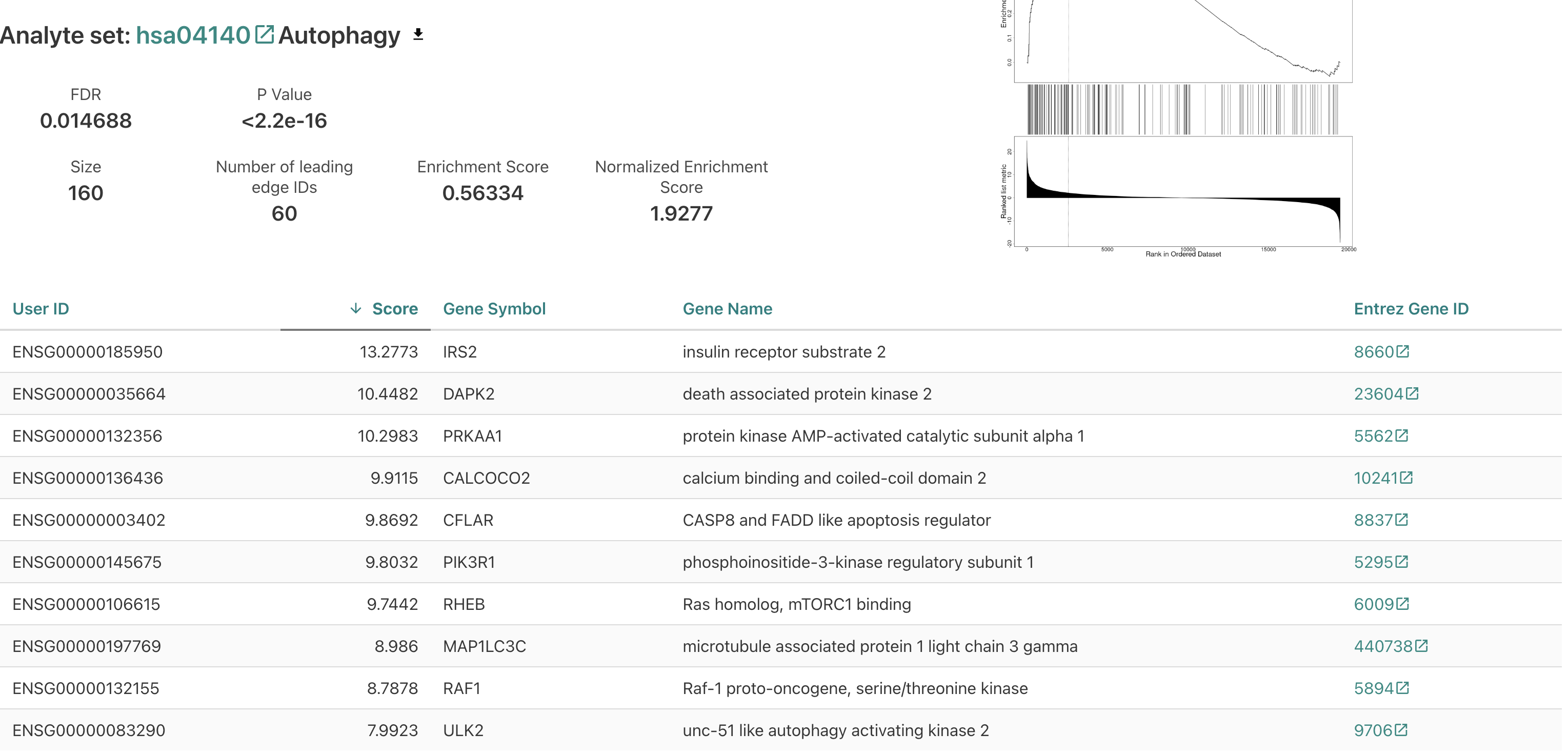
Nice! For this test we also get a value that gives the relative magnitude of change (here, all the categories seem to be ones associated with genes upregulated upon treatment).
Key Points
Normalization is necessary to account for different library sizes between samples.
Dimensional reduction (PCA) can help us understand the relationship between samples, and how it tracks with biological and technical variables.
We often want to include technical variables in the design formula, so that the differential expression comparison may include controlling for these as we solve for the effect of the biological variable.
Heatmaps are a useful visualization for understanding differential expression results, but must often be scaled by gene to become interpretable.
ORA and GSEA accept very different inputs (only DE genes versus all genes), and their outputs may be different as well as a result (though often they include similar categories).