Structural Variation in long reads
Overview
Teaching: 30 min
Exercises: 0 minQuestions
What are the advantages/disadvantages of long reads?
How might we leverage a combination of long and short read data?
Objectives
Investigate how long-read data can improve SV calling.
Long read platforms
The two major platforms in long read sequencing are PacBio and Oxford Nanopore.
PacBio’s flagship is the Revio, which produces reads in the 5kb to 35kb range with very high accuracy.

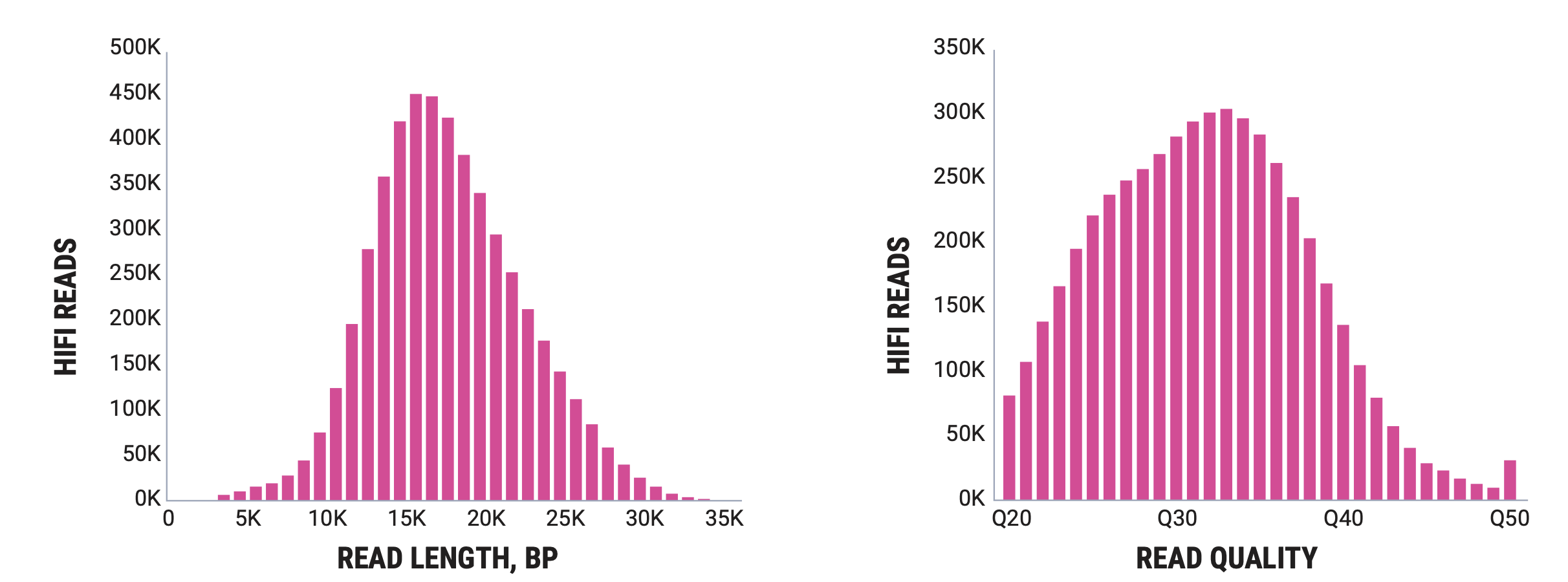
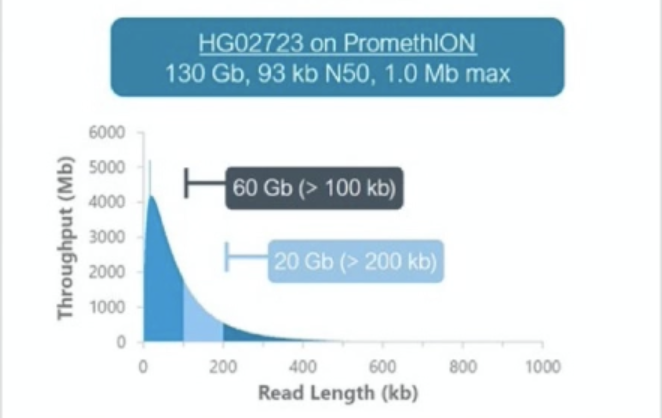
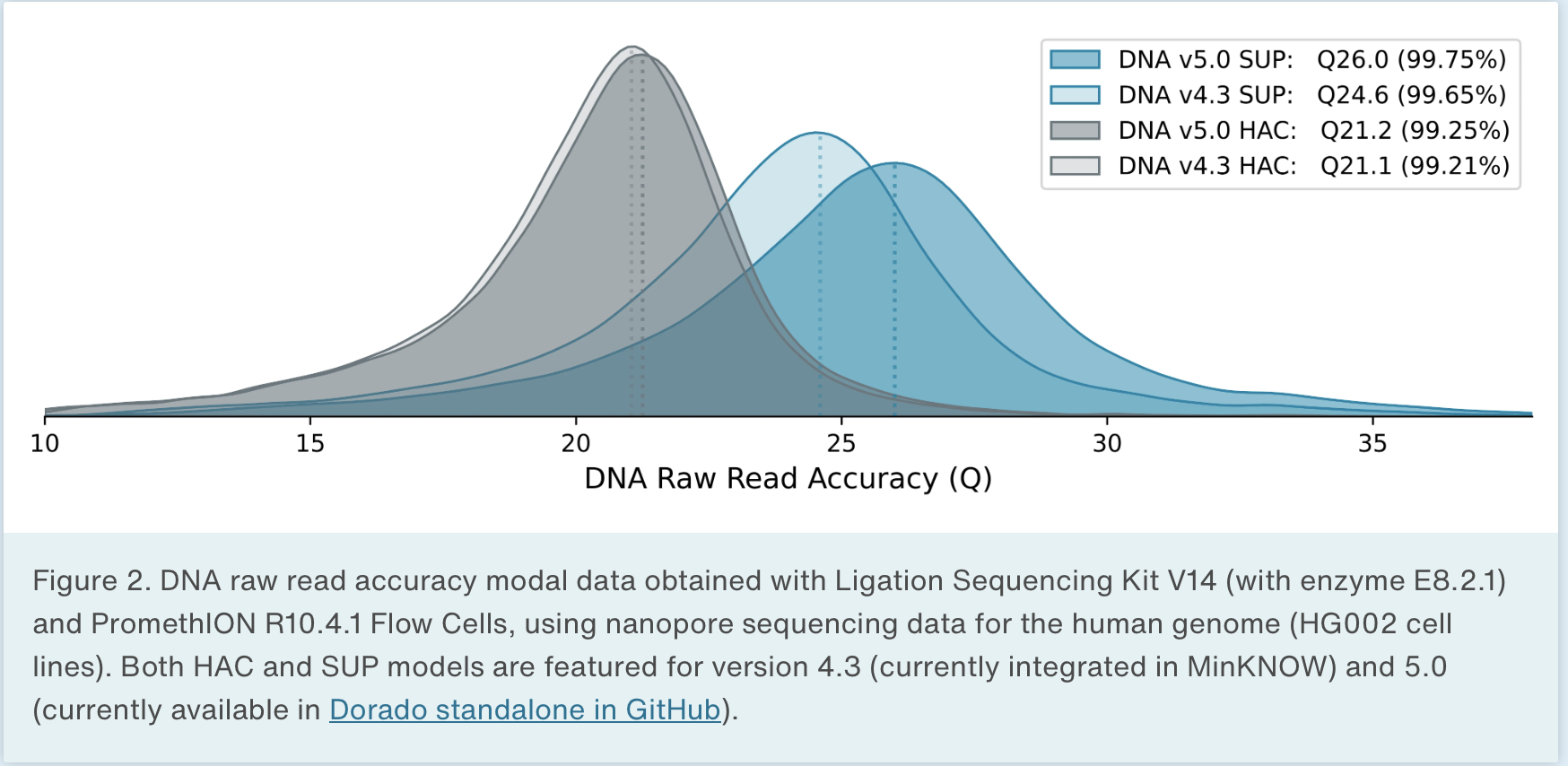
Oxford Nanopore produces sequencers that range in size from the MinION, which is roughly smart phone sized to the PromethION, the high throughput version that we have at NYGC. There are some differences in the read outputs of the various platforms but the MinION has been shown to produce N50 read lengths over 100kb with maximum read lengths greater than 800kb using ONT’s ultra-long sequencing prep. The PromethION can produce even greater N50 values and can produce megabase long reads. Typically these reads are lowe overall base quality than PacBio but ONT has steadily been improving the base quality for their data.


Advantages of long reads
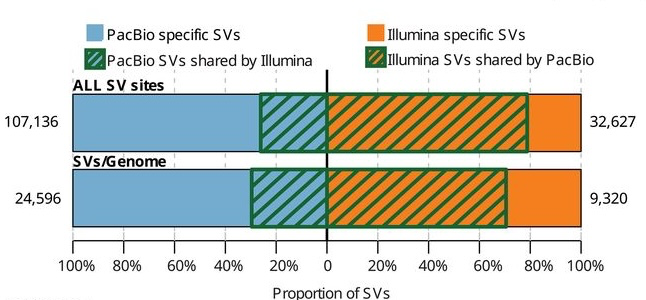
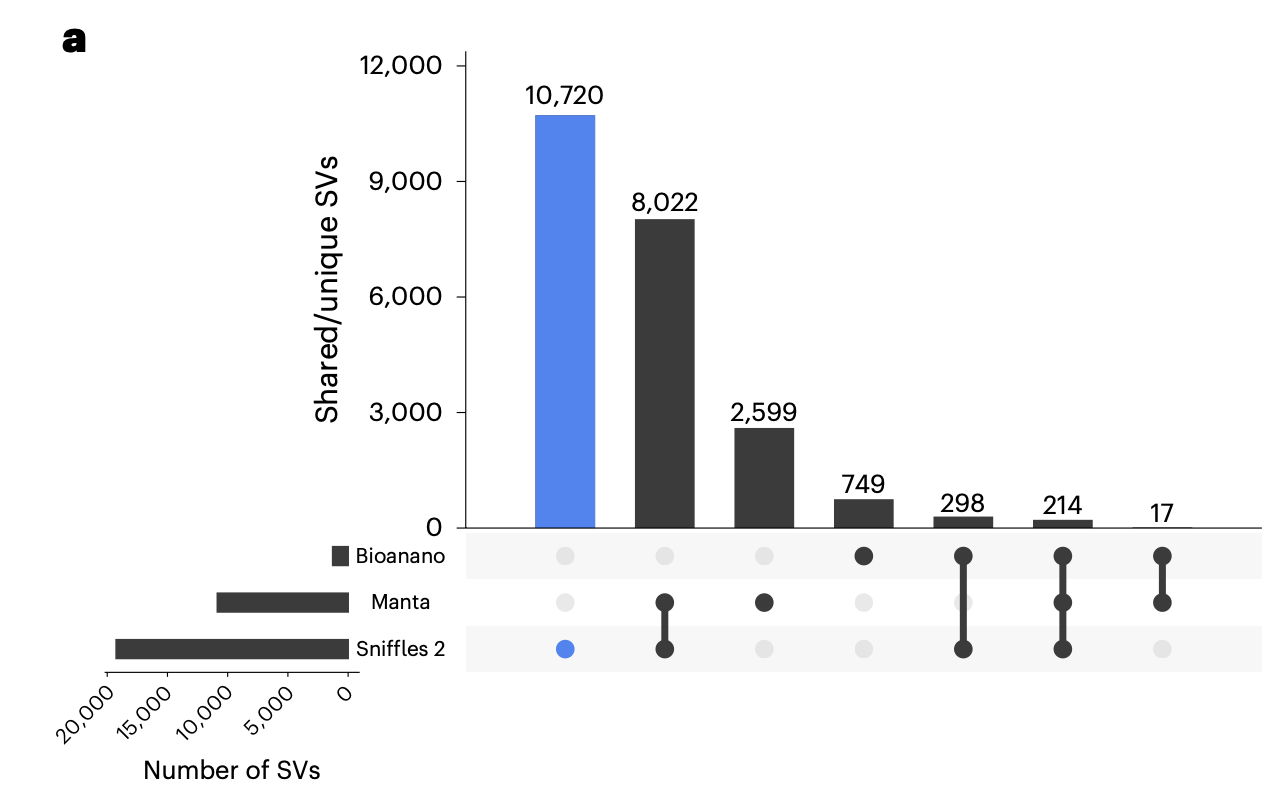
We can call many more SVs using long reads, either by alignment (with enough depth) or assembly (with sufficient assembly quality)1.
Challenge
Why are we able to call more variants with long reads? Why are there Illumina only SVs?
Solution
- Long reads can often be mapped to regions of the genome where short reads fail to map.
Long reads can span larger events making discovery easier and increasing breakpoint accuracy.
- False positives are one possible explaination for Illumina only SVs.
SV calling in long reads
Alignment
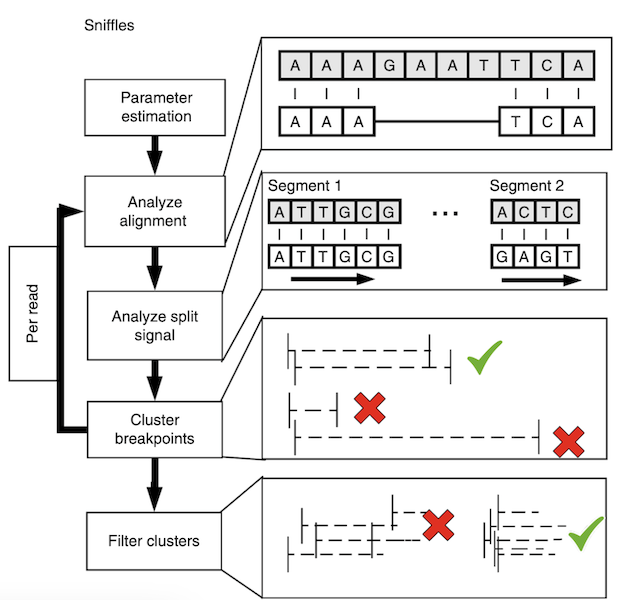
Sniffles uses a three step approach to calling SVs. First it scans the read alignments looking for split reads and inline events. Inline events are insertions and deletions that occur entirely within the read. It puts these SVs into bins and then looks for neighboring bins that can be merged using a repeat aware approach to create these clusters of SV candidates. Each cluster is then re-analyzed and a final determination is made based on read support, expected coverage changes and breakpoint variance.
Assembly
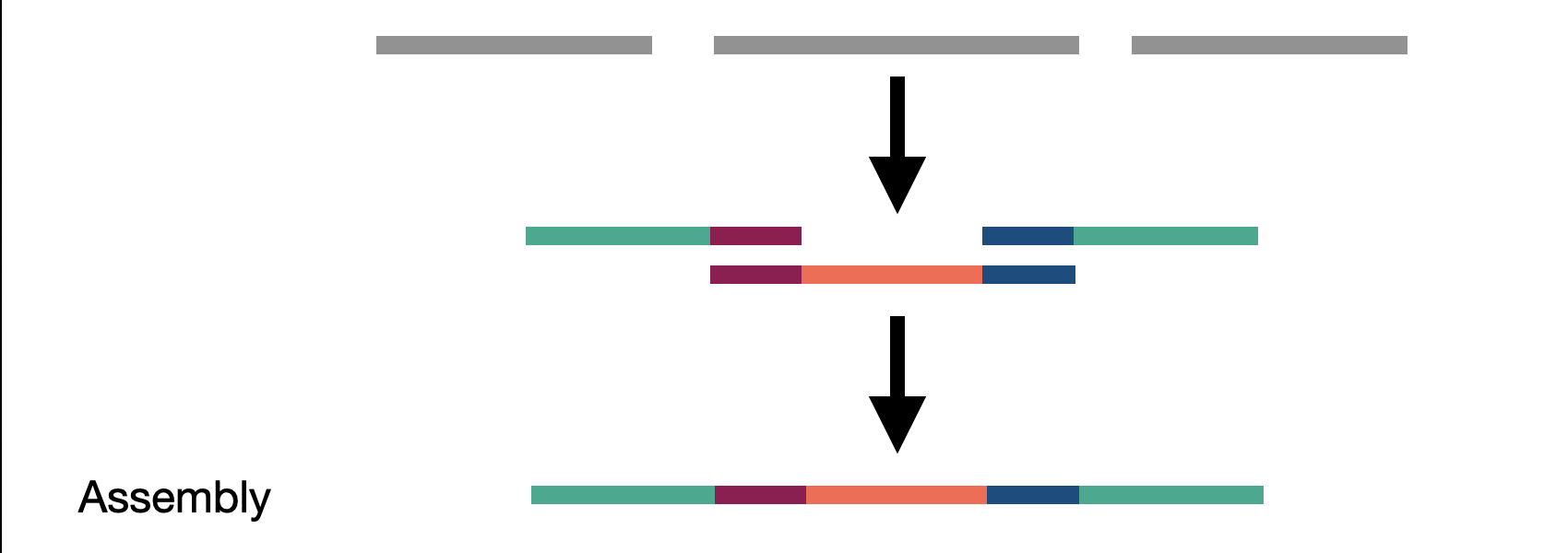
We touched on assembly in the short read section but here we actually refer to whole genome assembly compared to local assembly in short reads. By assembling as much of the genome as possible, including novel insertions, we create a bigger picture of our sample. These assembled fragments, called contigs, can then be aligned to the reference. The contigs act as a sort of ultra-long read as they represent many reads stiched together.
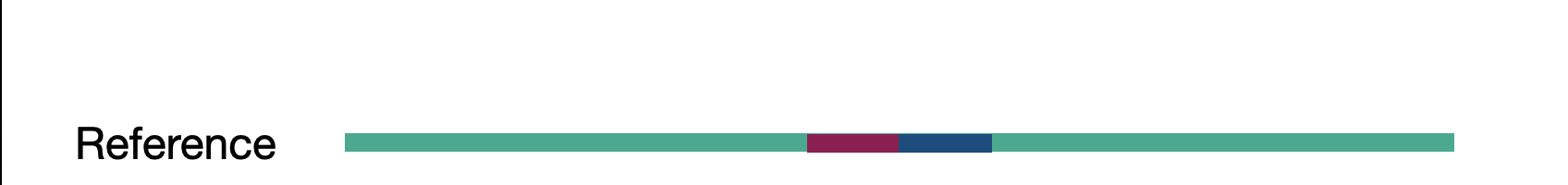
Challenge
Given the previous assembly result and reference sequence, what type of SV are we looking at here?
Drawbacks
- Long read sequencing is becoming more affordable but is still much more expensive than short read
sequencing
SR << LR Alignment << LR Assembly. - Throughput is lower, reducing the turn around time for projects with large numbers of samples.
- Sequencing prep, especially for ultra-long protocols is tedious and difficult to perform consitently.
- Due to cost, we typically don’t sequence samples at as high of depth.
- We are still limited to some extent by the length of our reads and our ablility to span an entire event with one or more reads and some regions of the genome are still very difficult to sequence and align to.
Genotyping LR SVs in SR data
Challenge
Given the section title, what two approaches might we take in creating a hybrid SV call set that uses both long and short reads?
Paragraph
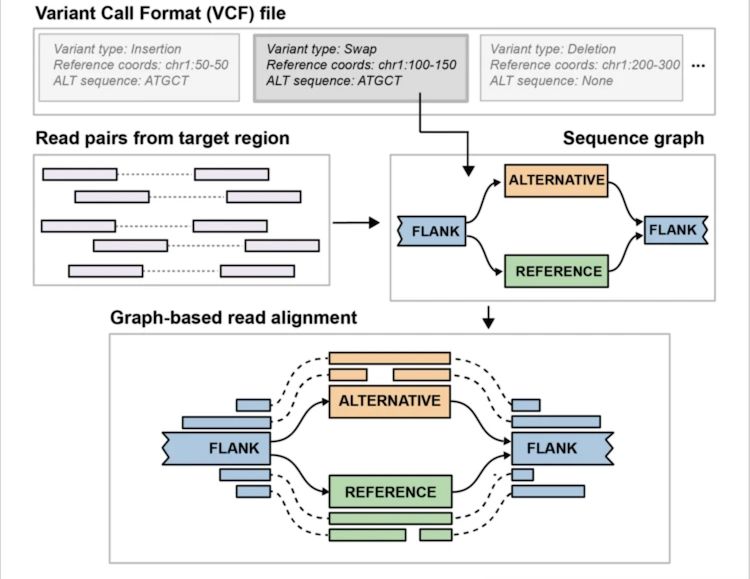
Pangenie
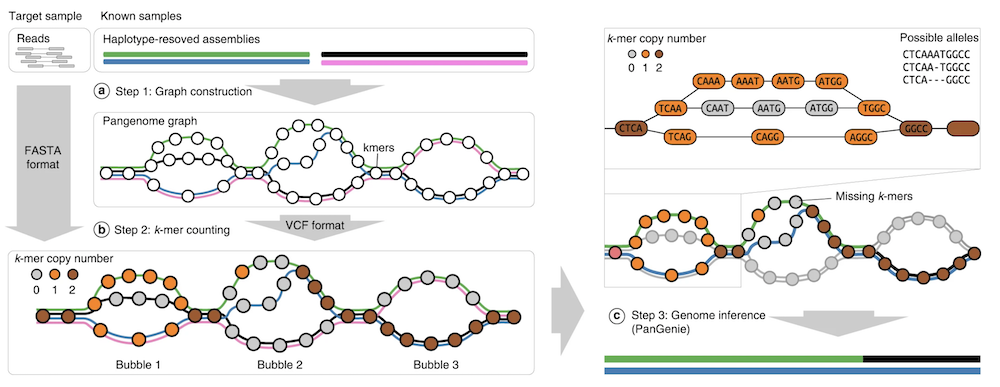
Solution
We can:
- Sequence a number of individuals with long reads and genotype those SV calls in our short read sample set.
- We can leverage existing long read SV callsets and genotype those SVs into out short read sample set.
Question
Does anyone know of any other technologies being used for structural variation?
Callback
Key Points
Long-reads offer significant advantages over short-reads for SV calling.
Genotyping of long-read discovered SVs in short-read data allows for some scalability.