Structural Variation in short reads
Overview
Teaching: 90 min
Exercises: 0 minQuestions
What is a structural variant?
Why is structural variantion important?
Objectives
Explain the difference between SNVs, INDELs, and SVs.
Explain the different types of SVs.
Explain the evidence we use to discover SVs.
Review
Simple read alignment

Simple InDel

What are structural variants
Structural variation is most typically defined as variation affecting larger fragments of the genome than SNVs and InDels; for our purposes those 50 base pairs or greater. This is an admittedly arbitrary definition, but it provides us a useful cutoff between InDels and SVs.
Importance of SVs
SVs affect an order of magnitude more bases in the human genome in comparison to SNVs (Pang et al, 2010) and are more likely to associate with disease.
Structural variation encompases several classes of variants including deletions, insertions, duplications, inversions, translocations, and copy number variations (CNVs). CNVs are a subset of structural variations, specifically deletions and duplications, that affect large (>10kb) segments of the genome.
Breakpoints
The term breakpoint is used to denote a boundry between a structural variation and the reference.
Examples
Deletion
Insertion
Duplication
Inversion
Translocation
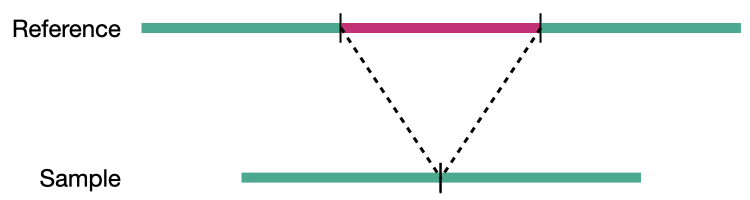
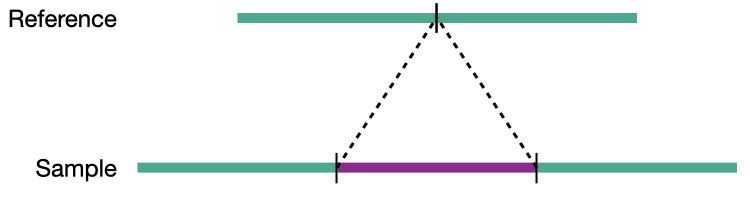
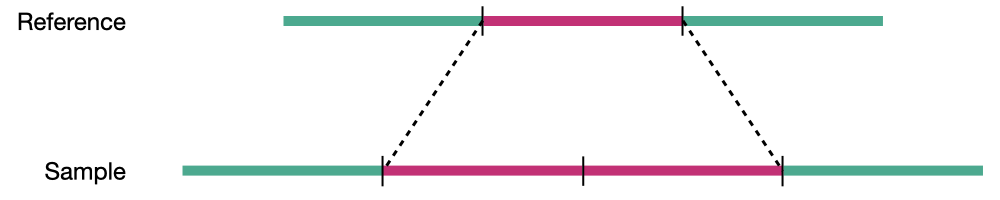
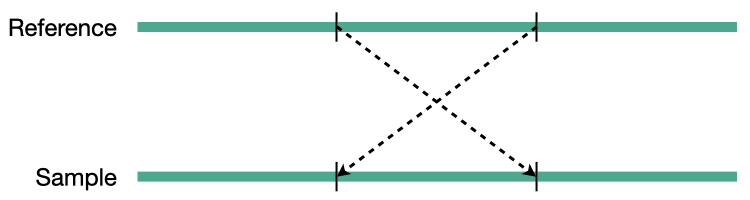
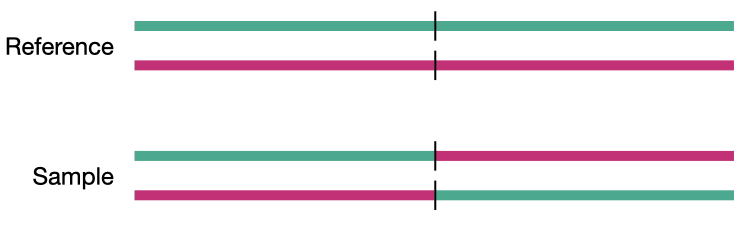
Detecting structural variants in short-read data
Because structural variants are most often larger than the individual reads we must use different types of read evidence than those used for SNVs and InDels which can be called by simple read alignment. We use three types of read evidence to discover structural variations: discordant read pairs, split-reads, and read depth.
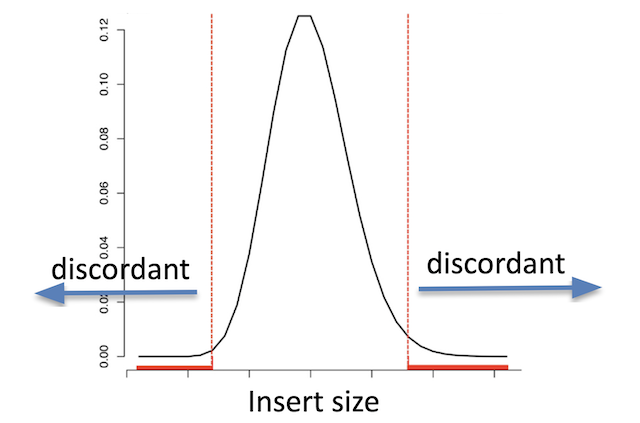
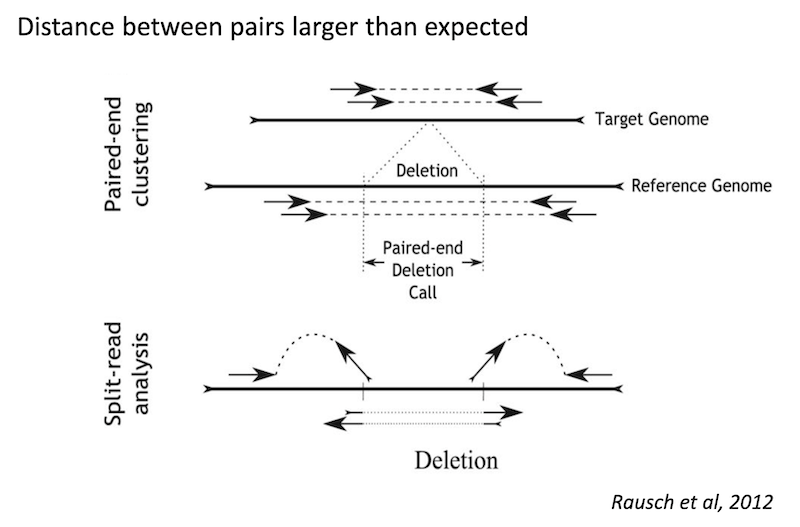
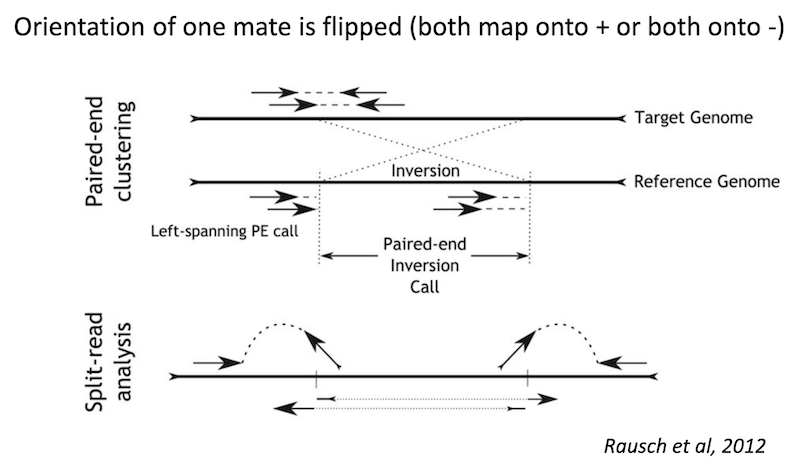
Discordant read pairs have insert sizes that fall significantly outside the normal distribution of insert sizes.
Insert size distribution
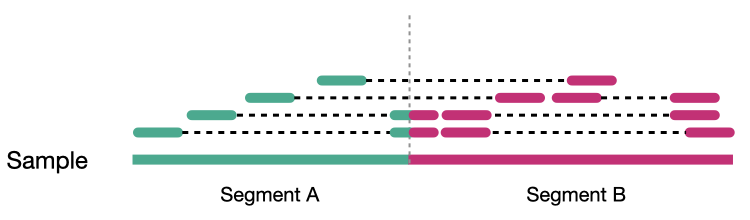
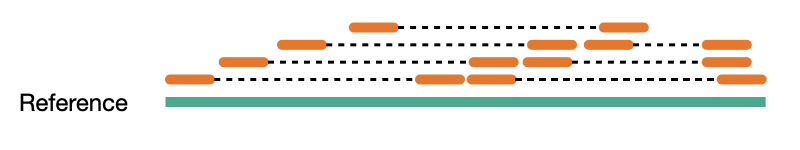
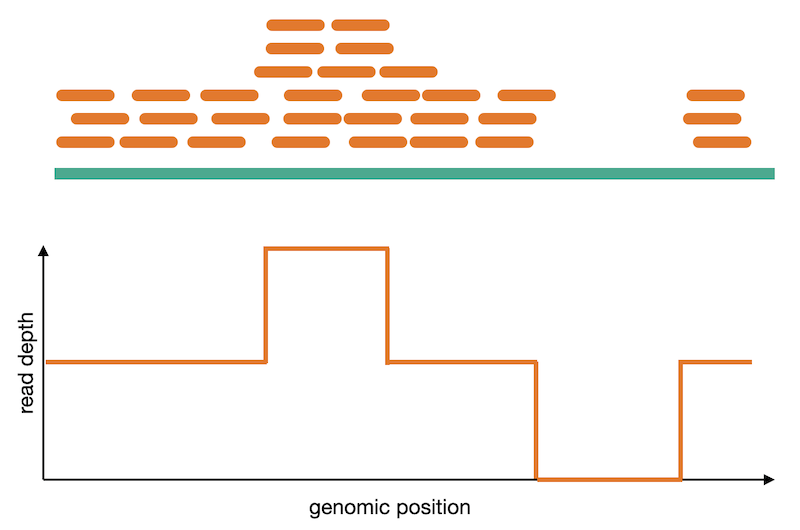
Split reads are those where part of the read aligns to the reference on one side of the breakpoint and the other part of the read aligns to the other side of the deletion breakpoint or to the inserted sequence. Read depth is where increases or decreases in read coverage occur versus the average read coverage of the genome.
Reads aligned to sample genome
Reads aligned to reference genome
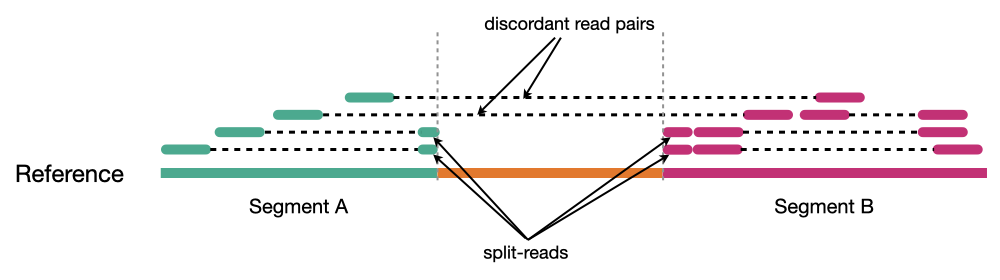
Coverage comes in two variants, sequence coverage and physical coverage. Sequence coverage is the number of times a base was read while physical coverage is the number of times a base was read or spanned by paired reads.
Sequence coverage

When there are no paired reads, sequence coverage equals the physical coverage. However, when paired reads are introduced the two coverage metrics can vary widely.
Physcial coverage
Read depth
Read signatures
Deletion read signature
Inversion read signature
Tandem duplication read signature
Translocation read signature
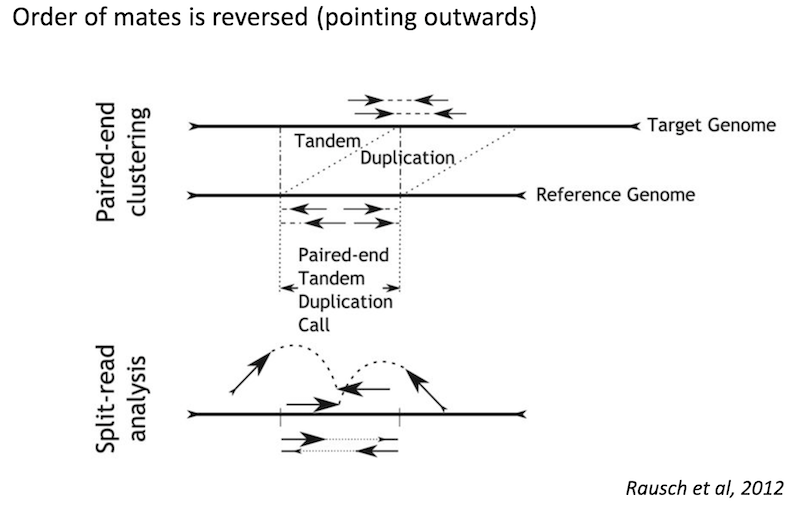
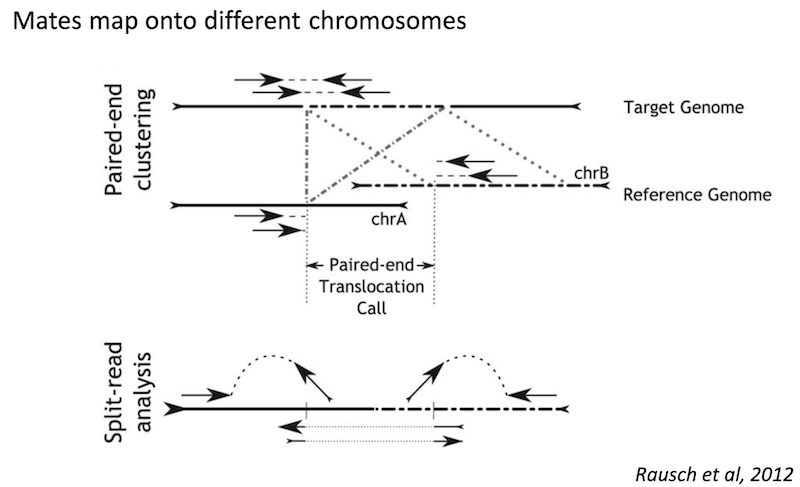
Challenge
What do you think the read signature of an insertion might look like?
Solution
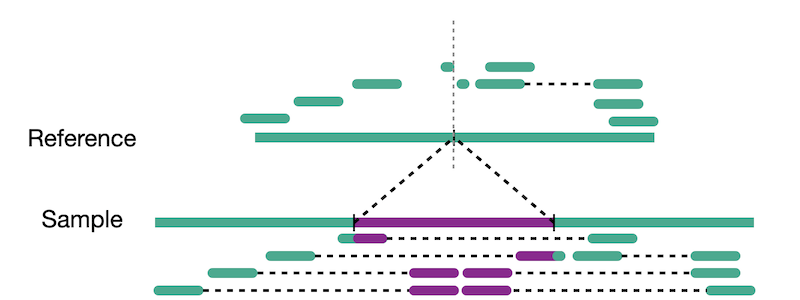
Copy number analysis
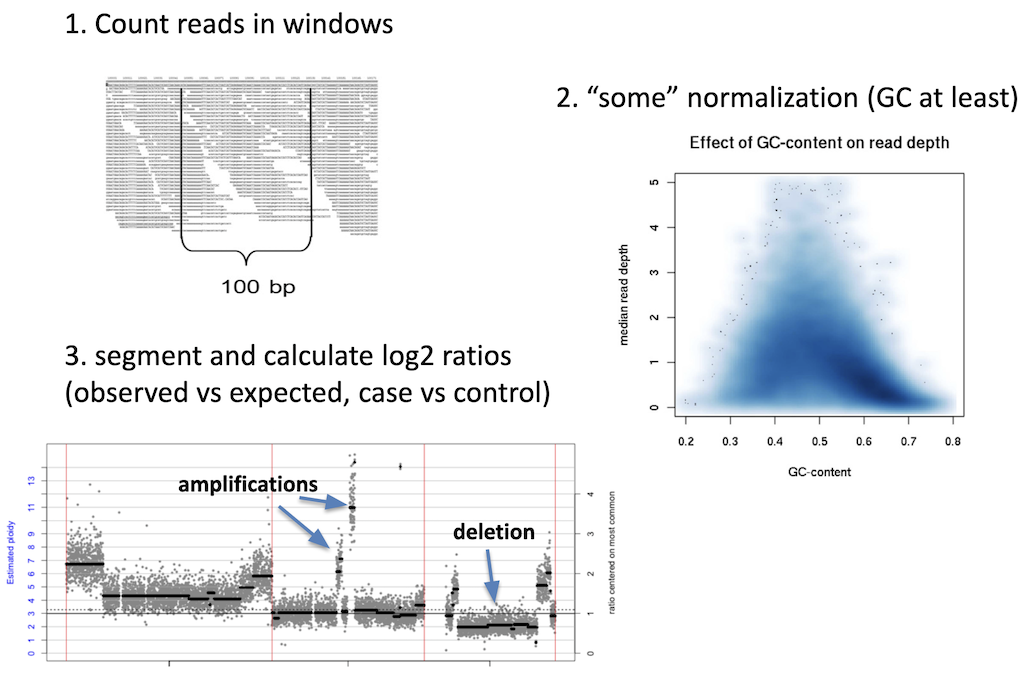
Calling of copy number variation from WGS data is done using read depth, where reads are counted in bins or windows across the entire genome. The counts need to have some normalization applied to them in order to account for sequencing irregularities such as mappability and GC content. These normalized counts can then be converted into their copy number equivalents using a process called segmentation. Read coverage is, however, inheirently noisy. It changes based on genomic regions, DNA quality, and other factors. This makes calling CNVs difficult and is why many CNV callers focus on large variants where it is easier to normalize away smaller confounding changes in read depth.
Caller resolution
We consider caller resolution to be how likely each algorithm is to determine the exact breakpoints of the SV. Precise location of SV breakpoints is an advantage when merging and regenotyping SVs. Here we are looking at the read signatures we’ve discussed so far: read depth, read pair, and split reads. We also see here another category which is assembly, which in this context means local assembly of the reads from the SV region is used to better determine the breakpoints of the SV.
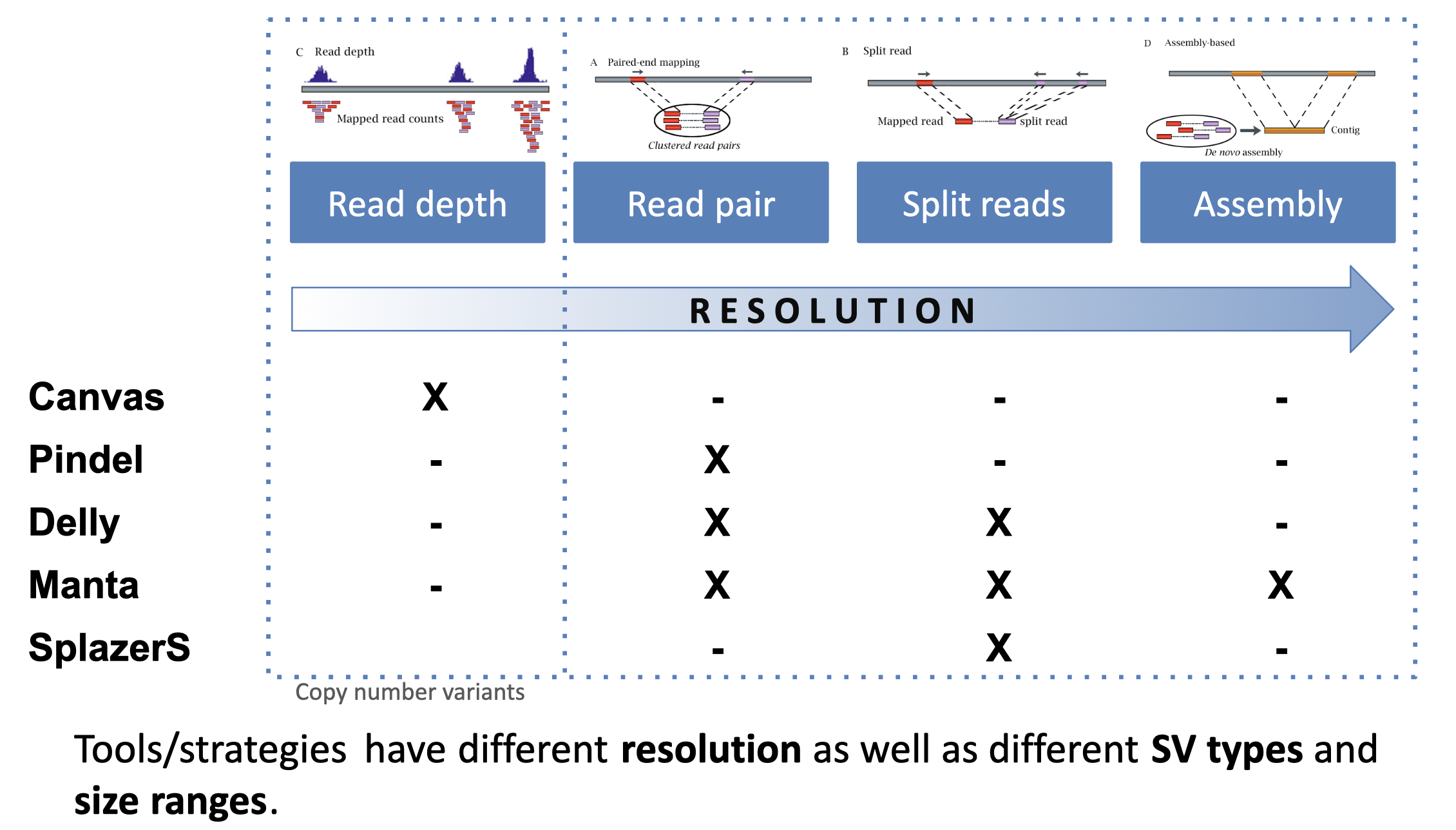
Caller concordance
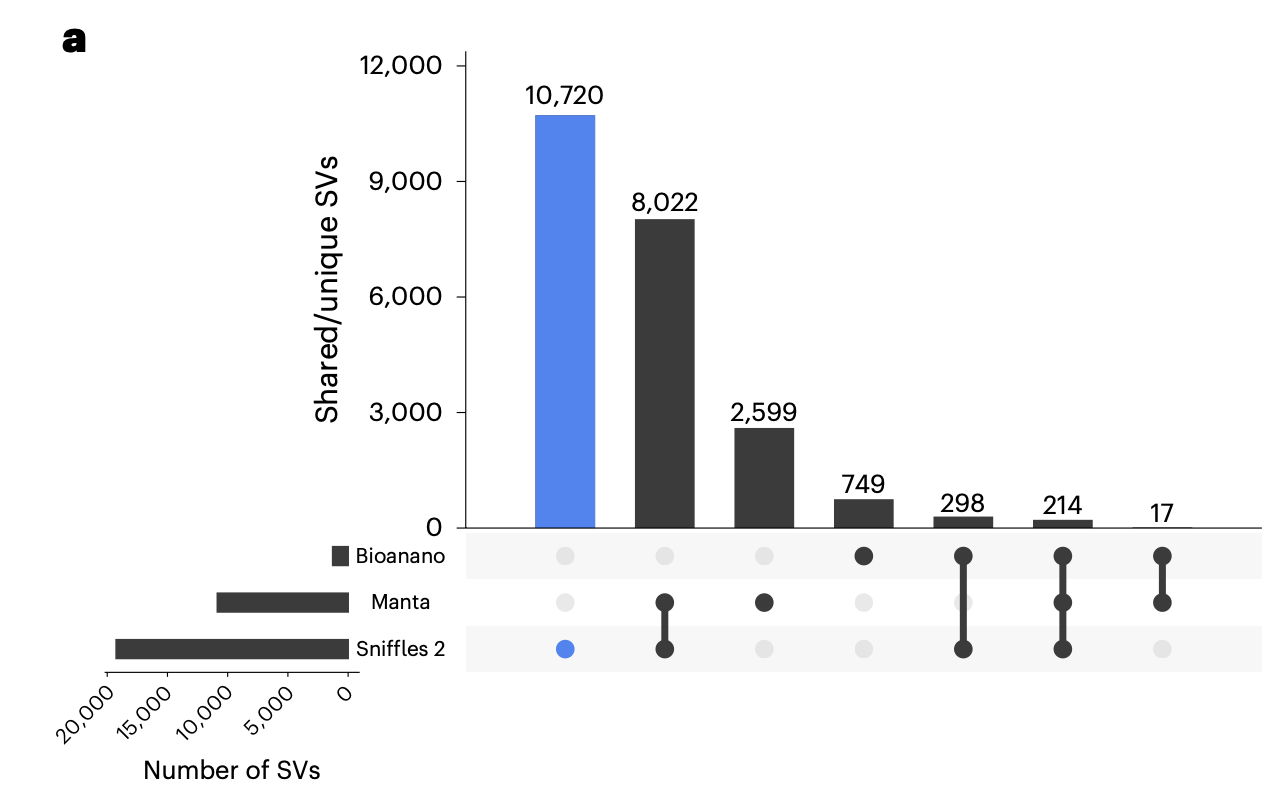
Because SV callers can both use different types of read evidence and apply different weights to the various read signatures, concordance between SV callers is usually quite low in comparison to SNV and InDel variant callers. Concordance between SV calls using different technologies show an even more pronounced lack of concordance.
Key Points
Structural variants are more difficult to identify.
Discovery of SVs usually requires multiple types of read evidence.
There is often significant disagreement between SV callers.